- STÉRÉOCHIMIE - Stéréochimie organique
- STÉRÉOCHIMIE - Stéréochimie organiqueOn trouvera dans l’article chimie ORGANIQUE le principe des méthodes qui ont conduit à l’établissement des formules développées planes. On va montrer ici pourquoi ces formules planes sont encore insuffisantes.Désignant provisoirement par X, Y, Z, T (X , Y , Z , T ) des substituants mono-atomiques tous différents, on est appelé à représenter par la formule plane (1) un dérivé tétrasubstitué du méthane, mais rien ne dit, a priori, si elle est équivalente à la formule (2).L’expérience montrant qu’un tel dérivé n’existe que sous une seule forme, on serait tenté de penser que l’ordre de disposition des substituants autour d’un même atome est indifférent, et de couper court à toute discussion en écrivant le composé CX2YZ.Mais, si l’on considère la formule à peine plus compliquée:
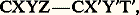 en admettant l’équivalence de toutes les formules planes, on devrait conclure qu’il lui correspond un composé unique; or, il en existe quatre. Il est donc de toute nécessité d’envisager la disposition spatiale des substituants autour d’un même atome. C’est l’objet de la stéréochimie organique.Cette discipline comprend deux parties: la stéréochimie statique , qui n’envisage que la structure spatiale des molécules, et la stéréochimie dynamique , qui étudie les variations des structures spatiales au cours des réactions.1. Stéréochimie statiqueCarbone «tétraédrique»Tout composé CX4, CX3Y, CX2Y2, CX2YZ existe sous une seule variété. Il faut, ou bien admettre que si un tel composé peut, à l’origine, prendre naissance sous différentes formes, toutes se transforment spontanément en le composé unique le plus stable, ou bien que les composés obtenus par différentes voies sont, dès l’origine, identiques. En généralisant la première hypothèse, on serait amené à nier l’existence de plusieurs isomères pour CXYZ 漣CX Y T , conclusion en opposition avec l’expérience. On a tenté de donner de la seconde hypothèse des démonstrations expérimentales; elles ne sont pas à l’abri de toute critique, et il est plus raisonnable de la considérer comme un postulat qui se vérifiera par toutes ses conséquences.L’unicité des composés CX4, CX3Y, CX2Y2, CX2YZ n’est compatible qu’avec une hypothèse structurale unique; dans CX4, les quatre X jouent un rôle identique puisqu’il n’existe qu’un composé CX3Y, et tout couple YZ est interchangeable avec le couple ZY, puisqu’il n’existe qu’un composé CX2YZ. Il faut donc que les quatre substituants identiques dans CX4 occupent les quatre sommets d’un tétraèdre régulier dont le carbone occupe le centre. Dans les composés CX3Y, CX2Y2, CX2YZ, ce tétraèdre n’est plus régulier, mais l’édifice conserve au moins un plan de symétrie, montrant que les dispositions (3) et (4) sont absolument équivalentes.Dans CX4, toutes les liaisons C 漣X ont même longueur, et les six angles de valence XCX ont même valeur:
en admettant l’équivalence de toutes les formules planes, on devrait conclure qu’il lui correspond un composé unique; or, il en existe quatre. Il est donc de toute nécessité d’envisager la disposition spatiale des substituants autour d’un même atome. C’est l’objet de la stéréochimie organique.Cette discipline comprend deux parties: la stéréochimie statique , qui n’envisage que la structure spatiale des molécules, et la stéréochimie dynamique , qui étudie les variations des structures spatiales au cours des réactions.1. Stéréochimie statiqueCarbone «tétraédrique»Tout composé CX4, CX3Y, CX2Y2, CX2YZ existe sous une seule variété. Il faut, ou bien admettre que si un tel composé peut, à l’origine, prendre naissance sous différentes formes, toutes se transforment spontanément en le composé unique le plus stable, ou bien que les composés obtenus par différentes voies sont, dès l’origine, identiques. En généralisant la première hypothèse, on serait amené à nier l’existence de plusieurs isomères pour CXYZ 漣CX Y T , conclusion en opposition avec l’expérience. On a tenté de donner de la seconde hypothèse des démonstrations expérimentales; elles ne sont pas à l’abri de toute critique, et il est plus raisonnable de la considérer comme un postulat qui se vérifiera par toutes ses conséquences.L’unicité des composés CX4, CX3Y, CX2Y2, CX2YZ n’est compatible qu’avec une hypothèse structurale unique; dans CX4, les quatre X jouent un rôle identique puisqu’il n’existe qu’un composé CX3Y, et tout couple YZ est interchangeable avec le couple ZY, puisqu’il n’existe qu’un composé CX2YZ. Il faut donc que les quatre substituants identiques dans CX4 occupent les quatre sommets d’un tétraèdre régulier dont le carbone occupe le centre. Dans les composés CX3Y, CX2Y2, CX2YZ, ce tétraèdre n’est plus régulier, mais l’édifice conserve au moins un plan de symétrie, montrant que les dispositions (3) et (4) sont absolument équivalentes.Dans CX4, toutes les liaisons C 漣X ont même longueur, et les six angles de valence XCX ont même valeur: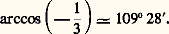 Dans les composés CX3Y, CX2Y2, CX2YZ, les distances interatomiques ne sont plus toutes égales et les angles de valence ne sont plus tous égaux; toutefois, tant qu’il s’agit de molécules acycliques, ces angles s’écartent peu de 1090 28 .Les structures réelles présentent les mêmes symétries qu’un tétraèdre régulier aux sommets duquel sont inscrites des lettres représentant les substituants. C’est pourquoi ce postulat est couramment désigné par «postulat du carbone tétraédrique». En réalité, ni les faces ni les arêtes de ce tétraèdre n’ont un rôle quelconque; seules les lignes de valence, axes de symétrie des orbitales, ont une signification physique et le postulat du carbone tétraédrique devrait s’énoncer: les axes des quatre orbitales du carbone ne sont pas dans un même plan, mais forment des angles égaux dans CX4, et différents dans CXYZT.La théorie de l’hybridation des orbitales est venue confirmer ce postulat, mais il est bien antérieur à cette théorie, et il a pu être formulé en s’appuyant exclusivement sur les considérations de symétrie; tout le reste de cet exposé reposera sur les mêmes méthodes.Carbone asymétriqueDans la représentation tétraédrique de CX2YZ, les deux X ne jouent pas le même rôle; en substituant l’un d’eux par T, le composé CXYZT ne présente plus d’élément de symétrie.Les deux formules (5) et (6) sont symétriques par rapport à un plan.Deux lots de molécules CXYZT, le premier exclusivement bâti sur le modèle (5), l’autre exclusivement bâti sur le modèle (6), auront des propriétés scalaires (densité, points d’ébullition et de fusion, etc.) identiques. Mais on ne peut affirmer qu’il en est de même pour les propriétés orientées. L’une d’elles est particulièrement caractéristique; il s’agit du pouvoir rotatoire naturel d’un composé à l’état liquide, ou en solution dans un solvant lui-même dépourvu de pouvoir rotatoire. Chacun des lots possède le pouvoir rotatoire naturel; de plus, les pouvoirs rotatoires des deux lots sont égaux et de sens contraires.On dit que (5) et (6) sont des inverses optiques ou des énantiomères ; l’un d’eux est dextrogyre, tandis que l’autre est lévogyre.L’expérience montre qu’un lot de composés de formule plane CXYZT a, ou n’a pas, le pouvoir rotatoire. Si le lot est uniquement composé de la forme (5) ou de la forme (6), il présente le pouvoir rotatoire maximal en valeur absolue; on dit que l’on a affaire à l’un des antipodes optiques purs. Si le lot est composé en quantités égales des formes (5) et (6), il n’a plus de pouvoir rotatoire; on le dit alors racémique (ou mélange racémique); enfin, un lot peut être constitué par un mélange en parties inégales de (5) et (6); il possède alors un pouvoir rotatoire de l’un ou de l’autre sens, mais inférieur en valeur absolue à celui des antipodes optiques; on a affaire à un mélange partiellement racémisé (ou partiellement déracémisé).En principe, tout racémique ou tout mélange racémisé peut être, par des méthodes appropriées, «dédoublé» en antipodes optiques purs. CXYZT est donc ou bien un antipode optique, ou bien un mélange de deux antipodes optiques.Le carbone lié à quatre substituants tous différents est appelé carbone asymétrique ; on lui affecte fréquemment un astérisque:
Dans les composés CX3Y, CX2Y2, CX2YZ, les distances interatomiques ne sont plus toutes égales et les angles de valence ne sont plus tous égaux; toutefois, tant qu’il s’agit de molécules acycliques, ces angles s’écartent peu de 1090 28 .Les structures réelles présentent les mêmes symétries qu’un tétraèdre régulier aux sommets duquel sont inscrites des lettres représentant les substituants. C’est pourquoi ce postulat est couramment désigné par «postulat du carbone tétraédrique». En réalité, ni les faces ni les arêtes de ce tétraèdre n’ont un rôle quelconque; seules les lignes de valence, axes de symétrie des orbitales, ont une signification physique et le postulat du carbone tétraédrique devrait s’énoncer: les axes des quatre orbitales du carbone ne sont pas dans un même plan, mais forment des angles égaux dans CX4, et différents dans CXYZT.La théorie de l’hybridation des orbitales est venue confirmer ce postulat, mais il est bien antérieur à cette théorie, et il a pu être formulé en s’appuyant exclusivement sur les considérations de symétrie; tout le reste de cet exposé reposera sur les mêmes méthodes.Carbone asymétriqueDans la représentation tétraédrique de CX2YZ, les deux X ne jouent pas le même rôle; en substituant l’un d’eux par T, le composé CXYZT ne présente plus d’élément de symétrie.Les deux formules (5) et (6) sont symétriques par rapport à un plan.Deux lots de molécules CXYZT, le premier exclusivement bâti sur le modèle (5), l’autre exclusivement bâti sur le modèle (6), auront des propriétés scalaires (densité, points d’ébullition et de fusion, etc.) identiques. Mais on ne peut affirmer qu’il en est de même pour les propriétés orientées. L’une d’elles est particulièrement caractéristique; il s’agit du pouvoir rotatoire naturel d’un composé à l’état liquide, ou en solution dans un solvant lui-même dépourvu de pouvoir rotatoire. Chacun des lots possède le pouvoir rotatoire naturel; de plus, les pouvoirs rotatoires des deux lots sont égaux et de sens contraires.On dit que (5) et (6) sont des inverses optiques ou des énantiomères ; l’un d’eux est dextrogyre, tandis que l’autre est lévogyre.L’expérience montre qu’un lot de composés de formule plane CXYZT a, ou n’a pas, le pouvoir rotatoire. Si le lot est uniquement composé de la forme (5) ou de la forme (6), il présente le pouvoir rotatoire maximal en valeur absolue; on dit que l’on a affaire à l’un des antipodes optiques purs. Si le lot est composé en quantités égales des formes (5) et (6), il n’a plus de pouvoir rotatoire; on le dit alors racémique (ou mélange racémique); enfin, un lot peut être constitué par un mélange en parties inégales de (5) et (6); il possède alors un pouvoir rotatoire de l’un ou de l’autre sens, mais inférieur en valeur absolue à celui des antipodes optiques; on a affaire à un mélange partiellement racémisé (ou partiellement déracémisé).En principe, tout racémique ou tout mélange racémisé peut être, par des méthodes appropriées, «dédoublé» en antipodes optiques purs. CXYZT est donc ou bien un antipode optique, ou bien un mélange de deux antipodes optiques.Le carbone lié à quatre substituants tous différents est appelé carbone asymétrique ; on lui affecte fréquemment un astérisque: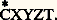 Depuis quelques années, on désigne par «chiralité» (du grec kheir , main) le fait, pour une molécule, de pouvoir exister sous deux formes inverses optiques, comme le sont (sensiblement) nos deux mains.Le carbone asymétrique est appelé «carbone chiral», ou «centre de chiralité»; il vaut mieux dire «site de chiralité», car on conçoit difficilement un «centre d’asymétrie».CX2YZ est un composé unique; si on effectue sur lui le remplacement expérimental de X par T (en n’utilisant que des réactifs symétriques), les probabilités pour obtenir (5) ou (6) sont égales et, comme de toute façon cette réaction ne peut être effectuée que sur un nombre considérable de molécules élémentaires, on conclut, d’après la loi des grands nombres, que le produit obtenu est pratiquement le racémique. C’est un cas particulier d’un principe beaucoup plus général, énoncé par Pasteur. Il est impossible d’aboutir à un composé actif sur la lumière polarisée en ne mettant en œuvre que des composés inactifs.Afin d’éviter des formules perspectives pour représenter des énantiomères, on est convenu de projeter les formules spatiales sur le plan du papier. On projette le tétraèdre sous forme d’un carré dont une diagonale est horizontale; les arêtes verticales et horizontales sont confondues sur cette projection avec les lignes de valence.On écrit les quatre substituants aux quatre sommets, formule (5). On passe de cette projection à celle de l’inverse optique (6) en permutant deux quelconques des lettres, mais, le plus souvent, on permute celles écrites verticalement.On supprime alors les quatre côtés du carré, on remplace le trait ponctué par un trait plein et on aboutit aux formules (7) et (8). Mais il faut bien préciser que les substituants écrits verticalement sont en avant, ceux écrits horizontalement sont en arrière du plan du papier; sinon les projections seraient ambiguës.Les formules projectives peuvent, sans changer la nature de l’énantiomère représenté, subir une rotation de 1800. (7) équivaut à (9), mais une rotation de 900 fait passer de l’un à l’autre: le composé représenté par la formule (10) est l’inverse optique de celui de la formule (7).On désigne par d la variété dextrogyre et par l la variété lévogyre, mais, dans la plupart des cas, on ignore la «configuration absolue»; on sait seulement que si (7) représente d , (10) représente l ; toutefois, dans des séries bien étudiées, la configuration absolue est connue; on représente alors par D une certaine configuration, et par L la configuration inverse, mais alors D n’est plus forcément le composé dextrogyre [cf. GLUCIDES].Il existe une nomenclature non ambiguë pour noter un carbone asymétrique dont la configuration absolue est connue.On établit un classement rationnel de tous les radicaux. Celui-ci fait intervenir, en priorité, le numéro atomique de l’atome d’attache du radical au carbone, puis, éventuellement, celui de ses substituants immédiats, puis, si besoin est, celui des substituants immédiats des précédents, etc.Voici quelques jalons de ce classement de substituants sur le carbone par priorité croissante:
Depuis quelques années, on désigne par «chiralité» (du grec kheir , main) le fait, pour une molécule, de pouvoir exister sous deux formes inverses optiques, comme le sont (sensiblement) nos deux mains.Le carbone asymétrique est appelé «carbone chiral», ou «centre de chiralité»; il vaut mieux dire «site de chiralité», car on conçoit difficilement un «centre d’asymétrie».CX2YZ est un composé unique; si on effectue sur lui le remplacement expérimental de X par T (en n’utilisant que des réactifs symétriques), les probabilités pour obtenir (5) ou (6) sont égales et, comme de toute façon cette réaction ne peut être effectuée que sur un nombre considérable de molécules élémentaires, on conclut, d’après la loi des grands nombres, que le produit obtenu est pratiquement le racémique. C’est un cas particulier d’un principe beaucoup plus général, énoncé par Pasteur. Il est impossible d’aboutir à un composé actif sur la lumière polarisée en ne mettant en œuvre que des composés inactifs.Afin d’éviter des formules perspectives pour représenter des énantiomères, on est convenu de projeter les formules spatiales sur le plan du papier. On projette le tétraèdre sous forme d’un carré dont une diagonale est horizontale; les arêtes verticales et horizontales sont confondues sur cette projection avec les lignes de valence.On écrit les quatre substituants aux quatre sommets, formule (5). On passe de cette projection à celle de l’inverse optique (6) en permutant deux quelconques des lettres, mais, le plus souvent, on permute celles écrites verticalement.On supprime alors les quatre côtés du carré, on remplace le trait ponctué par un trait plein et on aboutit aux formules (7) et (8). Mais il faut bien préciser que les substituants écrits verticalement sont en avant, ceux écrits horizontalement sont en arrière du plan du papier; sinon les projections seraient ambiguës.Les formules projectives peuvent, sans changer la nature de l’énantiomère représenté, subir une rotation de 1800. (7) équivaut à (9), mais une rotation de 900 fait passer de l’un à l’autre: le composé représenté par la formule (10) est l’inverse optique de celui de la formule (7).On désigne par d la variété dextrogyre et par l la variété lévogyre, mais, dans la plupart des cas, on ignore la «configuration absolue»; on sait seulement que si (7) représente d , (10) représente l ; toutefois, dans des séries bien étudiées, la configuration absolue est connue; on représente alors par D une certaine configuration, et par L la configuration inverse, mais alors D n’est plus forcément le composé dextrogyre [cf. GLUCIDES].Il existe une nomenclature non ambiguë pour noter un carbone asymétrique dont la configuration absolue est connue.On établit un classement rationnel de tous les radicaux. Celui-ci fait intervenir, en priorité, le numéro atomique de l’atome d’attache du radical au carbone, puis, éventuellement, celui de ses substituants immédiats, puis, si besoin est, celui des substituants immédiats des précédents, etc.Voici quelques jalons de ce classement de substituants sur le carbone par priorité croissante: Soit un carbone asymétrique dont les quatre substituants sont, dans l’ordre inverse du classement précédent, a, b, c, d. On place l’observateur à l’opposé de d. Si pour passer de a à b , puis à c , son regard se déplace dans le sens des aiguilles d’une montre, le carbone chiral est noté R (latin, rectus ); s’il se déplace dans l’autre sens, il est noté S (latin, sinister ). Un exemple en est donné à la figure.Principe de libre rotation (ou de liaison mobile)Deux carbones saturés simplement liés sont réunis par une seule ligne de valence, et chacun d’eux échange trois valences avec ses substituants. En général, les trois substituants de l’un des carbones peuvent tourner, par rapport à ceux de l’autre carbone, autour de la liaison C1 漣C2; mais les états de rotation relative ont des probabilités différentes: par exemple, dans le cas de CH2Br 漣CH2Br (bromure d’éthylène), un lot de telles molécules est constitué par une infinité de conformations. La moins probable est celle dans laquelle les deux atomes de carbone et les deux atomes de brome sont dans un même plan et ces derniers les plus proches l’un de l’autre (position antipode); la conformation la plus probable est celle dans laquelle ces quatre atomes sont dans le même plan, mais les atomes de brome le plus éloignés possible (position avantagée principale); toutefois, la probabilité de trouver la molécule dans une conformation déterminée ne varie pas de façon monotone avec l’angle des plans Br(1) C(1) C(2) et C(1) C(2) Br(2); il existe, d’une part, deux positions avantagées secondaires dans lesquelles Br(1), C(1), C(2), H(2) sont dans le même plan, H et Br étant le plus éloignés possible, et, d’autre part, deux positions antipodes secondaires dans lesquelles ils sont dans le même plan, mais le plus rapprochés possible.On représente ces positions par la projection de Newman ; la molécule est vue par un observateur placé dans le prolongement de C(2) 漣C(1) (cf. CONFORMATIONS [chimie], fig. 1). Les positions avantagées sont dites étoilées et les positions antipodes sont dites éclipsées. Dans le cas général les conformations éclipsées sont toujours les moins probables.Quoi qu’il en soit, au zéro absolu les molécules sont, pour la plupart, en position avantagée principale, en moindre concentration aux positions avantagées secondaires (qui constituent des «cuvettes d’énergie»). Lorsque la température s’élève, les molécules commencent par se disperser aux alentours immédiats des positions étoilées, puis, l’agitation thermique augmentant, elles franchissent les positions éclipsées (barrière d’énergie), conduisant ainsi à une libre rotation, mais avec une répartition statistique très différente des diverses conformations.Toutefois, aux températures accessibles, il est très rare que les molécules soient confinées dans les cuvettes d’énergie (cf. infra : Atropo-isomérie ). Dans l’immense majorité des cas, on peut admettre la libre rotation; en d’autres termes, les «isomères rotationnels» ne sont que très exceptionnellement séparables. On en conclut que le bromure d’éthylène, par exemple, s’il n’est pas constitué de molécules toutes identiques, forme du moins un lot unique et constitue en fait une seule espèce chimique ou, tout au moins, un «lot d’espèce unique».Pratiquement, presque toujours réalisable, la libre rotation permet de résoudre plusieurs problèmes stéréochimiques. Grâce à elle, on peut toujours imposer à une molécule ou à un radical ne renfermant pas de carbone asymétrique une conformation présentant un plan de symétrie.Cette remarque permet de lever la restriction faite au début. Tout ce qui a été dit reste vrai si X, Y, Z, T ne sont plus des substituants monoatomiques (présentant une symétrie de révolution), à condition qu’ils ne renferment pas de carbone asymétrique (ou un atome jouant le même rôle). En effet, si R, R , R ne renferment pas de carbone asymétrique, CRR (R )2 est susceptible de prendre une conformation d’ensemble douée d’un plan de symétrie, ce qui suffit pour conclure que CRR (R )2 est un lot d’espèce unique comme CXYZ2 et ne possède pas le pouvoir rotatoire.D’ailleurs, cette condition suffisante n’est pas nécessaire; le seul énoncé correct d’un principe attribué à Pasteur est celui-ci: la condition nécessaire et suffisante pour qu’un lot d’espèce unique ne possède pas le pouvoir rotatoire est qu’une conformation quelconque d’une molécule de ce lot puisse se transformer spontanément en son inverse optique. Ce passage peut, comme dans l’exemple précédent, se faire en franchissant une conformation symétrique, mais cette condition n’est pas nécessaire.Diastéréo-isomérieLorsqu’une molécule renferme plusieurs carbones asymétriques, on peut envisager, pour chacun d’eux, deux configurations différentes; il en résulte que, dans le cas général, à n carbones asymétriques correspondent 2n isomères auxquels on a donné le nom de «diastéréo-isomères». Toutefois, des symétries d’ensemble peuvent réduire ce nombre.Cas de deux carbones asymétriquesLe cas le plus simple est celui de deux carbones asymétriques voisins portant chacun trois substituants non respectivement identiques: CXYZ 漣CX Y Z , ou même CXYZ 漣CXYZ .On généralise la projection conventionnelle d’un seul carbone asymétrique: grâce au principe de liaison mobile, on amène Z, les deux C et Z à former une ligne polygonale ouverte, plane et convexe que l’on projette horizontalement, les deux C dans le plan du papier, les Z et Z en arrière. On obtient Z ... C 漣 C ... Z ou, plus simplement, Z 漣C 漣C 漣Z .On place alors X, Y, X , Y sur les carbones respectifs, en avant du plan de figure et de toutes les façons possibles, on obtient ainsi les formules (11), (12), (13) et (14). Les composés (11) et (12), ainsi que (13) et (14), sont inverses optiques; les autres couples possibles, non inverses optiques, sont dits diastéréo-isomères; les deux racémiques [(11) + (12)] et [(13) + (14)] sont dits racémiques diastéréo-isomères.Les diastéréo-isomères, contrairement aux inverses optiques, ont des propriétés différentes, ce qui permet, en principe du moins, de les séparer par les méthodes de l’analyse immédiate.Si X, Y, Z sont respectivement identiques à X , Y , Z , (13) et (14) restent actifs et inverses optiques; mais (11) et (12) sont identiques; comme ils sont également inverses optiques, les formules écrites ne sont que deux projections équivalentes (elles se déduisent d’ailleurs l’une de l’autre par une rotation permise de 1800) d’un composé unique inactif sur la lumière polarisée et désigné par «inactif indédoublable» pour le distinguer du racémique [(13) + (14)] qui est inactif mais dédoublable. (11) [ou (12)] est encore appelé isomère «méso» car il est intermédiaire entre (13) et (14).Dans la nomenclature se référant aux configurations absolues, le composé méso est noté R, S; le racémique est noté (R, R + S, S).Z et Z étant les substituants les plus compliqués, si X = X , que Y et Y soient identiques ou différents, on désigne par «érythro» les composés (11) et (12), et par «thréo» les composés (13) et (14), cela par référence à des oses: érythroses et thréoses présentant cette isomérie.La projection de l’inactif indédoublable, représentée par la formule (15), possède un axe de symétrie, trace d’un plan de symétrie de la conformation imposée; mais cette conformation (position antipode) est la moins probable; la position avantagée présente au contraire un centre de symétrie, mais ne peut être écrite en projection conventionnelle. Par contre, la projection conventionnelle de l’un des composés actifs, par exemple celui représenté par la formule (16), a un centre de symétrie; celui-ci ne supprime pas l’activité optique; ce n’est que la trace d’un axe de symétrie (qui, dans l’espace, n’implique pas l’absence de pouvoir rotatoire); ce n’est pas un centre de symétrie dans l’espace, car il ne peut être à la fois aux milieux des 2 C, des 2 X, des 2 Y et des 2 Z.Ces remarques sont généralisables aux projections des molécules présentant n carbones asymétriques incorporables dans une chaîne linéaire; dans le cas général, un axe de symétrie de la formule projective supprime l’activité optique; un centre de symétrie de cette formule ne le supprime pas.GénéralisationSi les carbones asymétriques font partie d’une chaîne linéaire, on peut généraliser ces projections; pour ce faire, on amène les n carbones asymétriques et l’un des substituants des carbones extrêmes à former une ligne polygonale plane et convexe; on la projette radialement sur un cylindre et on développe la projection pour obtenir:
Soit un carbone asymétrique dont les quatre substituants sont, dans l’ordre inverse du classement précédent, a, b, c, d. On place l’observateur à l’opposé de d. Si pour passer de a à b , puis à c , son regard se déplace dans le sens des aiguilles d’une montre, le carbone chiral est noté R (latin, rectus ); s’il se déplace dans l’autre sens, il est noté S (latin, sinister ). Un exemple en est donné à la figure.Principe de libre rotation (ou de liaison mobile)Deux carbones saturés simplement liés sont réunis par une seule ligne de valence, et chacun d’eux échange trois valences avec ses substituants. En général, les trois substituants de l’un des carbones peuvent tourner, par rapport à ceux de l’autre carbone, autour de la liaison C1 漣C2; mais les états de rotation relative ont des probabilités différentes: par exemple, dans le cas de CH2Br 漣CH2Br (bromure d’éthylène), un lot de telles molécules est constitué par une infinité de conformations. La moins probable est celle dans laquelle les deux atomes de carbone et les deux atomes de brome sont dans un même plan et ces derniers les plus proches l’un de l’autre (position antipode); la conformation la plus probable est celle dans laquelle ces quatre atomes sont dans le même plan, mais les atomes de brome le plus éloignés possible (position avantagée principale); toutefois, la probabilité de trouver la molécule dans une conformation déterminée ne varie pas de façon monotone avec l’angle des plans Br(1) C(1) C(2) et C(1) C(2) Br(2); il existe, d’une part, deux positions avantagées secondaires dans lesquelles Br(1), C(1), C(2), H(2) sont dans le même plan, H et Br étant le plus éloignés possible, et, d’autre part, deux positions antipodes secondaires dans lesquelles ils sont dans le même plan, mais le plus rapprochés possible.On représente ces positions par la projection de Newman ; la molécule est vue par un observateur placé dans le prolongement de C(2) 漣C(1) (cf. CONFORMATIONS [chimie], fig. 1). Les positions avantagées sont dites étoilées et les positions antipodes sont dites éclipsées. Dans le cas général les conformations éclipsées sont toujours les moins probables.Quoi qu’il en soit, au zéro absolu les molécules sont, pour la plupart, en position avantagée principale, en moindre concentration aux positions avantagées secondaires (qui constituent des «cuvettes d’énergie»). Lorsque la température s’élève, les molécules commencent par se disperser aux alentours immédiats des positions étoilées, puis, l’agitation thermique augmentant, elles franchissent les positions éclipsées (barrière d’énergie), conduisant ainsi à une libre rotation, mais avec une répartition statistique très différente des diverses conformations.Toutefois, aux températures accessibles, il est très rare que les molécules soient confinées dans les cuvettes d’énergie (cf. infra : Atropo-isomérie ). Dans l’immense majorité des cas, on peut admettre la libre rotation; en d’autres termes, les «isomères rotationnels» ne sont que très exceptionnellement séparables. On en conclut que le bromure d’éthylène, par exemple, s’il n’est pas constitué de molécules toutes identiques, forme du moins un lot unique et constitue en fait une seule espèce chimique ou, tout au moins, un «lot d’espèce unique».Pratiquement, presque toujours réalisable, la libre rotation permet de résoudre plusieurs problèmes stéréochimiques. Grâce à elle, on peut toujours imposer à une molécule ou à un radical ne renfermant pas de carbone asymétrique une conformation présentant un plan de symétrie.Cette remarque permet de lever la restriction faite au début. Tout ce qui a été dit reste vrai si X, Y, Z, T ne sont plus des substituants monoatomiques (présentant une symétrie de révolution), à condition qu’ils ne renferment pas de carbone asymétrique (ou un atome jouant le même rôle). En effet, si R, R , R ne renferment pas de carbone asymétrique, CRR (R )2 est susceptible de prendre une conformation d’ensemble douée d’un plan de symétrie, ce qui suffit pour conclure que CRR (R )2 est un lot d’espèce unique comme CXYZ2 et ne possède pas le pouvoir rotatoire.D’ailleurs, cette condition suffisante n’est pas nécessaire; le seul énoncé correct d’un principe attribué à Pasteur est celui-ci: la condition nécessaire et suffisante pour qu’un lot d’espèce unique ne possède pas le pouvoir rotatoire est qu’une conformation quelconque d’une molécule de ce lot puisse se transformer spontanément en son inverse optique. Ce passage peut, comme dans l’exemple précédent, se faire en franchissant une conformation symétrique, mais cette condition n’est pas nécessaire.Diastéréo-isomérieLorsqu’une molécule renferme plusieurs carbones asymétriques, on peut envisager, pour chacun d’eux, deux configurations différentes; il en résulte que, dans le cas général, à n carbones asymétriques correspondent 2n isomères auxquels on a donné le nom de «diastéréo-isomères». Toutefois, des symétries d’ensemble peuvent réduire ce nombre.Cas de deux carbones asymétriquesLe cas le plus simple est celui de deux carbones asymétriques voisins portant chacun trois substituants non respectivement identiques: CXYZ 漣CX Y Z , ou même CXYZ 漣CXYZ .On généralise la projection conventionnelle d’un seul carbone asymétrique: grâce au principe de liaison mobile, on amène Z, les deux C et Z à former une ligne polygonale ouverte, plane et convexe que l’on projette horizontalement, les deux C dans le plan du papier, les Z et Z en arrière. On obtient Z ... C 漣 C ... Z ou, plus simplement, Z 漣C 漣C 漣Z .On place alors X, Y, X , Y sur les carbones respectifs, en avant du plan de figure et de toutes les façons possibles, on obtient ainsi les formules (11), (12), (13) et (14). Les composés (11) et (12), ainsi que (13) et (14), sont inverses optiques; les autres couples possibles, non inverses optiques, sont dits diastéréo-isomères; les deux racémiques [(11) + (12)] et [(13) + (14)] sont dits racémiques diastéréo-isomères.Les diastéréo-isomères, contrairement aux inverses optiques, ont des propriétés différentes, ce qui permet, en principe du moins, de les séparer par les méthodes de l’analyse immédiate.Si X, Y, Z sont respectivement identiques à X , Y , Z , (13) et (14) restent actifs et inverses optiques; mais (11) et (12) sont identiques; comme ils sont également inverses optiques, les formules écrites ne sont que deux projections équivalentes (elles se déduisent d’ailleurs l’une de l’autre par une rotation permise de 1800) d’un composé unique inactif sur la lumière polarisée et désigné par «inactif indédoublable» pour le distinguer du racémique [(13) + (14)] qui est inactif mais dédoublable. (11) [ou (12)] est encore appelé isomère «méso» car il est intermédiaire entre (13) et (14).Dans la nomenclature se référant aux configurations absolues, le composé méso est noté R, S; le racémique est noté (R, R + S, S).Z et Z étant les substituants les plus compliqués, si X = X , que Y et Y soient identiques ou différents, on désigne par «érythro» les composés (11) et (12), et par «thréo» les composés (13) et (14), cela par référence à des oses: érythroses et thréoses présentant cette isomérie.La projection de l’inactif indédoublable, représentée par la formule (15), possède un axe de symétrie, trace d’un plan de symétrie de la conformation imposée; mais cette conformation (position antipode) est la moins probable; la position avantagée présente au contraire un centre de symétrie, mais ne peut être écrite en projection conventionnelle. Par contre, la projection conventionnelle de l’un des composés actifs, par exemple celui représenté par la formule (16), a un centre de symétrie; celui-ci ne supprime pas l’activité optique; ce n’est que la trace d’un axe de symétrie (qui, dans l’espace, n’implique pas l’absence de pouvoir rotatoire); ce n’est pas un centre de symétrie dans l’espace, car il ne peut être à la fois aux milieux des 2 C, des 2 X, des 2 Y et des 2 Z.Ces remarques sont généralisables aux projections des molécules présentant n carbones asymétriques incorporables dans une chaîne linéaire; dans le cas général, un axe de symétrie de la formule projective supprime l’activité optique; un centre de symétrie de cette formule ne le supprime pas.GénéralisationSi les carbones asymétriques font partie d’une chaîne linéaire, on peut généraliser ces projections; pour ce faire, on amène les n carbones asymétriques et l’un des substituants des carbones extrêmes à former une ligne polygonale plane et convexe; on la projette radialement sur un cylindre et on développe la projection pour obtenir: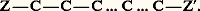 On place, de toutes les façons possibles, les deux autres substituants de chaque carbone. Si la formule plane ne présente pas un plan de symétrie, le nombre d’isomères est toujours 2n ; s’il en présente un, ce nombre est réduit à 2n -1 si n est impair et à 2n -1 + 2(n/2 )-1 s’il est pair. À titre d’exemple, nous citerons les alcools CH2OH 漣(CHOH)n 漣CH2OH; nous utiliserons une représentation simplifiée qui symbolise les composés de formules (17) et (18) par R 漣 face="EU Caron" ゲ 漣 R et R 漣r 漣 R .À la formule plane CH2OH 漣(CHOH)3 漣CH2OH correspondent quatre isomères:
On place, de toutes les façons possibles, les deux autres substituants de chaque carbone. Si la formule plane ne présente pas un plan de symétrie, le nombre d’isomères est toujours 2n ; s’il en présente un, ce nombre est réduit à 2n -1 si n est impair et à 2n -1 + 2(n/2 )-1 s’il est pair. À titre d’exemple, nous citerons les alcools CH2OH 漣(CHOH)n 漣CH2OH; nous utiliserons une représentation simplifiée qui symbolise les composés de formules (17) et (18) par R 漣 face="EU Caron" ゲ 漣 R et R 漣r 漣 R .À la formule plane CH2OH 漣(CHOH)3 漣CH2OH correspondent quatre isomères: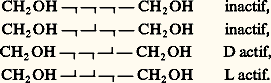 Pour CH2OH 漣(CHOH)4 漣CH2OH, on prévoit (et on connaît) dix isomères dont deux inactifs, et quatre couples de racémiques ; pour CH2OH 漣(CHOH)5 漣CH2OH, on prévoit seize isomères dont quatre inactifs, et six couples de racémiques; pour CH2OH 漣(CHOH)6 漣CH2OH, on prévoit trente-six isomères dont quatre inactifs, et seize couples de racémiques, etc.Si les carbones asymétriques ne peuvent être incorporés dans une chaîne linéaire, les projections conventionnelles ne sont plus d’aucun secours; ainsi, à la formule (19) correspondent 25 = 32 isomères. Mais la formule (20) n’en représente plus que 5.Si on symbolise par d et l les deux configurations possibles de 漣CXYZ, on peut les schématiser Cd 4, Cd 3l , Cd 2l 2, Cdl 3, Cl 4; tous sont actifs, sauf le troisième; Cd 2l 2 ne peut acquérir, quelle que soit sa conformation, un centre ou un plan de symétrie, mais il existe des conformations superposables à leur image, condition suffisante mais encore non nécessaire à l’inactivité optique.Stabilité et conformation des cyclanes et de leurs dérivésLes liaisons entre les carbones constituant un cycle à n atomes forment un angle au moins égal à l’angle au sommet du polygone régulier de n côtés.Pour n = 3, cet angle est de 600, soit très inférieur à l’angle normal (1090 28 ) de deux liaisons du carbone; il en résulte une «tension» du cycle qui explique sa grande instabilité.Pour n = 4, si les carbones sont dans un même plan, cet angle est de 900; la tension est encore notable; elle peut même augmenter très légèrement si le cycle subit un gauchissement.Pour n = 5, l’angle est de 1080; dans le cycle plan, la tension est négligeable.Pour n 礪 5, si le cycle était plan et convexe, on serait tenté d’envisager une tension de sens inverse de celle constatée pour les petits cycles (pour n = 6, 1200 au lieu de 1090 28 , et pour n très grand, 1800 au lieu de 1090 28 ). Or, on constate que pour n 閭 5 tous les cycles présentent une grande stabilité; on en conclut que pour n 礪 5 les cycles ne sont plus plans. En effet, pour n = 6, on prévoit deux dispositions sans tension. La première, la plus stable, est généralement désignée sous l’expression de forme chaise (cf. CONFORMATIONS) représentée en (21); elle n’est pas déformable sans augmentation transitoire des tensions. La seconde, en général moins stable, est appelée forme bateau (22); elle peut subir des déformations limitées, sans variation des tensions, en des conformations ne présentant plus de symétrie (formes croisées).En ce qui concerne le cyclohexane, il est en perpétuel équilibre entre deux formes chaise équivalentes (23) et une forme bateau déformable: à l’équilibre à la température ordinaire, la seconde ne représente que quelques millièmes de l’ensemble mais, grâce à cet équilibre pour lequel on peut envisager un passage par la forme plane, les isomères conformationnels constituent un lot d’espèce unique. C’est la raison pour laquelle, dans l’immense majorité des cas, on ne peut prévoir pour les dérivés de substitution du cyclohexane un nombre d’isomères supérieur à celui qui se déduirait d’une formule plane. La même remarque est valable pour les cycles à plus de six atomes de carbone.Décompte des isomères cyclaniquesLes cyclanes fondamentaux ou polyméthylènes (CH2)n n’existent que sous une seule forme; il en est de même des dérivés monosubstitués ou bisubstitués géminés dont la conformation plane présente un plan de symétrie.Par contre, l’introduction d’un second substituant, si ce n’est pas sur le carbone diamétralement opposé au carbone substitué, crée, d’un seul coup, deux carbones asymétriques, c’est-à-dire l’existence de quatre isomères deux à deux inverses optiques (24).Toutefois, si X et Y sont identiques, d 1 et l 1 ne représentent qu’un seul et même composé, un inactif indédoublable.d 1 et l 1, les composés «érythro», sont, ici, appelés isomères cis (les 2 H sont du même côté du plan moyen du cycle); d 2 et l 2, les composés «thréo», sont appelés isomères trans ; les racémiques (d 1 + l 1), (d 2 + l 2) sont, respectivement, les racémiques cis et trans .Évidemment, l’isomérie est la même si les carbones substitués ne sont plus consécutifs, pourvu – si le cycle est pair – qu’ils ne soient pas diamétralement opposés; s’il en est ainsi, apparaît une nouvelle isomérie.Les deux composés (25) et (26) sont l’un et l’autre uniques et inactifs, la figure présentant un plan de symétrie, mais ils sont différents l’un de l’autre; il existe donc un dérivé cis et un dérivé trans , l’un et l’autre dépourvus de pouvoir rotatoire; doués de propriétés physiques différentes, ils sont, en principe, séparables par les méthodes de l’analyse immédiate.Si le cycle est plus substitué, on peut prévoir un plus grand nombre d’isomères; il suffira de donner un exemple. Aux cyclohexanehexols de formule (27) correspondent neuf isomères, dont sept inactifs, et un couple de racémiques; tous sont connus; rappelons, à titre de comparaison, que CH3 漣(CHOH)6 漣CH3 représente trente-six isomères.Conformation des cycles hexagonauxSi l’on considère la forme chaise, généralement la plus stable, on remarque que les liaisons émanant du cycle sont pour six d’entre elles (1, 2, 3, 4, 5, 6) sensiblement dans le plan moyen du cycle, tandis que les six autres (7, 8, 9, 10, 11, 12) sont sensiblement parallèles entre elles et perpendiculaires à ce plan moyen (28). Les premières s’appellent liaisons équatoriales (e ), et les secondes liaisons axiales (a ).Pour le cyclohexane, la forme chaise est unique ou, plus exactement, les deux formes chaise que l’on peut écrire sont équivalentes: il n’en est plus de même pour un cyclohexane substitué; on peut en effet imaginer deux formes chaise différentes (29) et (30). Elles n’ont pas la même probabilité; en effet, dans la seconde, X se trouve, dans l’espace, très proche de deux hydrogènes axiaux explicités, ce qui provoque une gêne stérique et une instabilité; cette gêne stérique est d’autant plus importante que X est plus volumineux, d’où la règle générale : l’importance à l’équilibre de l’isomère où X occupe la position équatoriale est d’autant plus grande que X est plus volumineux; pratiquement, le tertiobutyl cyclohexane, X = C(CH3)3, est intégralement sous cette forme.S’il existe plusieurs substituants, ils tendent à occuper, si c’est possible, les positions équatoriales pour lesquelles la gêne stérique est à peu près nulle; c’est ainsi que les dérivés trans 見 (31) ou 塚 (32) ont cette conformation privilégiée. Il en est de même pour les dérivés cis 廓 (33).Par contre, pour les dérivés cis 見 ou 塚, et pour les dérivés trans 廓, il y a ambiguïté; en général, le substituant le plus volumineux occupe la position équatoriale. Mais si X et Y sont l’un et l’autre très volumineux, la forme bateau (ou croisée) devient la plus stable: exemple, le cis ditertiobutyl-cyclohexane-1,4 (34).Les contrôles physiques, en particulier la résonance magnétique protonique, ont confirmé l’exactitude de ces prévisions.Polycycles accolésDeux cycles pentagonaux accolés par deux carbones sont sensiblement plans, mais les plans des deux cycles forment un angle notable. Deux cycles pentagonaux ayant trois carbones en commun sont équivalents à un cycle hexagonal «ponté» par un atome de carbone; le cycle hexagonal prend alors obligatoirement la forme bateau.Deux cycles hexagonaux accolés par deux carbones peuvent, sous forme chaise, présenter les configurations (35) et (36).Dans l’hydrogénation poussée du naphtalène, il se forme simultanément les deux isomères qui ne sont pas interconvertibles.Certains composés bicycliques (37) comportent deux carbones asymétriques. On serait tenté de prévoir 22 = 4 isomères, deux à deux inverses optiques; il n’en est rien, car les deux carbones asymétriques ne sont pas indépendants; la configuration de l’un impose celle de l’autre, de sorte qu’il n’existe que deux isomères inverses optiques; la formule (38), bien que comportant deux carbones asymétriques, ne représente qu’un composé unique, car elle possède un plan de symétrie.De toute façon, dans le décompte des isomères possibles, il faut considérer l’indépendance des carbones asymétriques.Isomérie «cis-trans» éthylénique (ou géométrique)La liaison éthylénique peut être assimilée à un «cycle à deux atomes» (ce cycle est constitué par l’orbitale 神). En conséquence, l’isomérie des éthyléniques substitués est la même que celle des cycles pairs substitués sur des carbones diamétralement opposés; c’est ainsi qu’à la formule CHX=CHY correspondent deux isomères (39) et (40).Ces composés sont inactifs, la molécule présentant, comme plan de symétrie, le plan de la feuille de papier, car C,C,X,Y,H,H sont coplanaires; ils sont différents et ne subissent que difficilement une interconversion. En d’autres termes, le principe de libre rotation n’est pas applicable à la liaison éthylénique.Les formules conventionnelles (39) et (40) ne doivent pas être mélangées à celles du carbone asymétrique; on prévoirait ainsi sept isomères pour:
Pour CH2OH 漣(CHOH)4 漣CH2OH, on prévoit (et on connaît) dix isomères dont deux inactifs, et quatre couples de racémiques ; pour CH2OH 漣(CHOH)5 漣CH2OH, on prévoit seize isomères dont quatre inactifs, et six couples de racémiques; pour CH2OH 漣(CHOH)6 漣CH2OH, on prévoit trente-six isomères dont quatre inactifs, et seize couples de racémiques, etc.Si les carbones asymétriques ne peuvent être incorporés dans une chaîne linéaire, les projections conventionnelles ne sont plus d’aucun secours; ainsi, à la formule (19) correspondent 25 = 32 isomères. Mais la formule (20) n’en représente plus que 5.Si on symbolise par d et l les deux configurations possibles de 漣CXYZ, on peut les schématiser Cd 4, Cd 3l , Cd 2l 2, Cdl 3, Cl 4; tous sont actifs, sauf le troisième; Cd 2l 2 ne peut acquérir, quelle que soit sa conformation, un centre ou un plan de symétrie, mais il existe des conformations superposables à leur image, condition suffisante mais encore non nécessaire à l’inactivité optique.Stabilité et conformation des cyclanes et de leurs dérivésLes liaisons entre les carbones constituant un cycle à n atomes forment un angle au moins égal à l’angle au sommet du polygone régulier de n côtés.Pour n = 3, cet angle est de 600, soit très inférieur à l’angle normal (1090 28 ) de deux liaisons du carbone; il en résulte une «tension» du cycle qui explique sa grande instabilité.Pour n = 4, si les carbones sont dans un même plan, cet angle est de 900; la tension est encore notable; elle peut même augmenter très légèrement si le cycle subit un gauchissement.Pour n = 5, l’angle est de 1080; dans le cycle plan, la tension est négligeable.Pour n 礪 5, si le cycle était plan et convexe, on serait tenté d’envisager une tension de sens inverse de celle constatée pour les petits cycles (pour n = 6, 1200 au lieu de 1090 28 , et pour n très grand, 1800 au lieu de 1090 28 ). Or, on constate que pour n 閭 5 tous les cycles présentent une grande stabilité; on en conclut que pour n 礪 5 les cycles ne sont plus plans. En effet, pour n = 6, on prévoit deux dispositions sans tension. La première, la plus stable, est généralement désignée sous l’expression de forme chaise (cf. CONFORMATIONS) représentée en (21); elle n’est pas déformable sans augmentation transitoire des tensions. La seconde, en général moins stable, est appelée forme bateau (22); elle peut subir des déformations limitées, sans variation des tensions, en des conformations ne présentant plus de symétrie (formes croisées).En ce qui concerne le cyclohexane, il est en perpétuel équilibre entre deux formes chaise équivalentes (23) et une forme bateau déformable: à l’équilibre à la température ordinaire, la seconde ne représente que quelques millièmes de l’ensemble mais, grâce à cet équilibre pour lequel on peut envisager un passage par la forme plane, les isomères conformationnels constituent un lot d’espèce unique. C’est la raison pour laquelle, dans l’immense majorité des cas, on ne peut prévoir pour les dérivés de substitution du cyclohexane un nombre d’isomères supérieur à celui qui se déduirait d’une formule plane. La même remarque est valable pour les cycles à plus de six atomes de carbone.Décompte des isomères cyclaniquesLes cyclanes fondamentaux ou polyméthylènes (CH2)n n’existent que sous une seule forme; il en est de même des dérivés monosubstitués ou bisubstitués géminés dont la conformation plane présente un plan de symétrie.Par contre, l’introduction d’un second substituant, si ce n’est pas sur le carbone diamétralement opposé au carbone substitué, crée, d’un seul coup, deux carbones asymétriques, c’est-à-dire l’existence de quatre isomères deux à deux inverses optiques (24).Toutefois, si X et Y sont identiques, d 1 et l 1 ne représentent qu’un seul et même composé, un inactif indédoublable.d 1 et l 1, les composés «érythro», sont, ici, appelés isomères cis (les 2 H sont du même côté du plan moyen du cycle); d 2 et l 2, les composés «thréo», sont appelés isomères trans ; les racémiques (d 1 + l 1), (d 2 + l 2) sont, respectivement, les racémiques cis et trans .Évidemment, l’isomérie est la même si les carbones substitués ne sont plus consécutifs, pourvu – si le cycle est pair – qu’ils ne soient pas diamétralement opposés; s’il en est ainsi, apparaît une nouvelle isomérie.Les deux composés (25) et (26) sont l’un et l’autre uniques et inactifs, la figure présentant un plan de symétrie, mais ils sont différents l’un de l’autre; il existe donc un dérivé cis et un dérivé trans , l’un et l’autre dépourvus de pouvoir rotatoire; doués de propriétés physiques différentes, ils sont, en principe, séparables par les méthodes de l’analyse immédiate.Si le cycle est plus substitué, on peut prévoir un plus grand nombre d’isomères; il suffira de donner un exemple. Aux cyclohexanehexols de formule (27) correspondent neuf isomères, dont sept inactifs, et un couple de racémiques; tous sont connus; rappelons, à titre de comparaison, que CH3 漣(CHOH)6 漣CH3 représente trente-six isomères.Conformation des cycles hexagonauxSi l’on considère la forme chaise, généralement la plus stable, on remarque que les liaisons émanant du cycle sont pour six d’entre elles (1, 2, 3, 4, 5, 6) sensiblement dans le plan moyen du cycle, tandis que les six autres (7, 8, 9, 10, 11, 12) sont sensiblement parallèles entre elles et perpendiculaires à ce plan moyen (28). Les premières s’appellent liaisons équatoriales (e ), et les secondes liaisons axiales (a ).Pour le cyclohexane, la forme chaise est unique ou, plus exactement, les deux formes chaise que l’on peut écrire sont équivalentes: il n’en est plus de même pour un cyclohexane substitué; on peut en effet imaginer deux formes chaise différentes (29) et (30). Elles n’ont pas la même probabilité; en effet, dans la seconde, X se trouve, dans l’espace, très proche de deux hydrogènes axiaux explicités, ce qui provoque une gêne stérique et une instabilité; cette gêne stérique est d’autant plus importante que X est plus volumineux, d’où la règle générale : l’importance à l’équilibre de l’isomère où X occupe la position équatoriale est d’autant plus grande que X est plus volumineux; pratiquement, le tertiobutyl cyclohexane, X = C(CH3)3, est intégralement sous cette forme.S’il existe plusieurs substituants, ils tendent à occuper, si c’est possible, les positions équatoriales pour lesquelles la gêne stérique est à peu près nulle; c’est ainsi que les dérivés trans 見 (31) ou 塚 (32) ont cette conformation privilégiée. Il en est de même pour les dérivés cis 廓 (33).Par contre, pour les dérivés cis 見 ou 塚, et pour les dérivés trans 廓, il y a ambiguïté; en général, le substituant le plus volumineux occupe la position équatoriale. Mais si X et Y sont l’un et l’autre très volumineux, la forme bateau (ou croisée) devient la plus stable: exemple, le cis ditertiobutyl-cyclohexane-1,4 (34).Les contrôles physiques, en particulier la résonance magnétique protonique, ont confirmé l’exactitude de ces prévisions.Polycycles accolésDeux cycles pentagonaux accolés par deux carbones sont sensiblement plans, mais les plans des deux cycles forment un angle notable. Deux cycles pentagonaux ayant trois carbones en commun sont équivalents à un cycle hexagonal «ponté» par un atome de carbone; le cycle hexagonal prend alors obligatoirement la forme bateau.Deux cycles hexagonaux accolés par deux carbones peuvent, sous forme chaise, présenter les configurations (35) et (36).Dans l’hydrogénation poussée du naphtalène, il se forme simultanément les deux isomères qui ne sont pas interconvertibles.Certains composés bicycliques (37) comportent deux carbones asymétriques. On serait tenté de prévoir 22 = 4 isomères, deux à deux inverses optiques; il n’en est rien, car les deux carbones asymétriques ne sont pas indépendants; la configuration de l’un impose celle de l’autre, de sorte qu’il n’existe que deux isomères inverses optiques; la formule (38), bien que comportant deux carbones asymétriques, ne représente qu’un composé unique, car elle possède un plan de symétrie.De toute façon, dans le décompte des isomères possibles, il faut considérer l’indépendance des carbones asymétriques.Isomérie «cis-trans» éthylénique (ou géométrique)La liaison éthylénique peut être assimilée à un «cycle à deux atomes» (ce cycle est constitué par l’orbitale 神). En conséquence, l’isomérie des éthyléniques substitués est la même que celle des cycles pairs substitués sur des carbones diamétralement opposés; c’est ainsi qu’à la formule CHX=CHY correspondent deux isomères (39) et (40).Ces composés sont inactifs, la molécule présentant, comme plan de symétrie, le plan de la feuille de papier, car C,C,X,Y,H,H sont coplanaires; ils sont différents et ne subissent que difficilement une interconversion. En d’autres termes, le principe de libre rotation n’est pas applicable à la liaison éthylénique.Les formules conventionnelles (39) et (40) ne doivent pas être mélangées à celles du carbone asymétrique; on prévoirait ainsi sept isomères pour: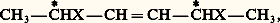 alors qu’il n’y en a que six: un cis inactif, un cis droit, un cis gauche, un trans inactif, un trans droit, un trans gauche; le cumul des deux conventions laisserait supposer que le trans inactif est un racémique, car sa projection ne peut présenter un axe de symétrie, mais, dans l’espace, une de ses conformations possède un centre de symétrie.Isomérie spiranniqueOn appelle spiranne une molécule possédant un seul carbone commun à deux cycles. Les plans moyens de ces cycles sont perpendiculaires; si les cycles sont pairs et si on introduit, dans chacun d’eux, un substituant sur le carbone diamétralement opposé au carbone commun, la formule (41) ne comporte aucun carbone asymétrique, sensu stricto .Mais les vecteurs HXet HYsont perpendiculaires dans l’espace; ils peuvent être soit dextrorsomes, soit sinistrorsomes, et les deux configurations sont différentes et non superposables mais inverses optiques: en conclusion, à la formule (41) correspondent deux énantiomères.Le même raisonnement est valable pour les composés (42) et (43), qui sont effectivement dédoublables en inverses optiques.On appelle parfois carbone «crypto-asymétrique» le carbone commun aux deux cycles, ou le carbone commun à un cycle et à une double liaison, ou encore le carbone commun à deux doubles liaisons.Atropo-isomérieL’existence d’un seul carbone asymétrique ou cryptoasymétrique dans une molécule est une condition suffisante à l’existence des énantiomères; on a cru longtemps qu’elle était nécessaire; il n’en est rien.Le noyau benzénique est plan et les six liaisons qui en émanent sont dans ce plan et forment, deux à deux, des angles de 600.Le diphényle est l’hydrocarbure (44). Il est sensiblement plan, la mésomérie se transmettant d’un noyau à l’autre et donnant une grande stabilité à la structure dans laquelle les deux noyaux sont coplanaires. Cependant, cette planéité n’est pas rigoureuse, les hydrogènes explicités se gênant légèrement; il n’en est plus de même si nous substituons à ces hydrogènes des radicaux volumineux; dans un tel composé, les plans de deux noyaux forment un angle notable, et les molécules s’écartent peu de la position dans laquelle ils sont perpendiculaires.Soit maintenant un dérivé (45) trisubstitué, avec X et Y différents. L’encombrement dû à X, Y, Z entrave complètement la libre rotation autour de la liaison qui unit les noyaux. Il s’ensuit que si nous supposons le noyau de gauche dans le plan de figure, le noyau de droite oscille peu de part et d’autre de la conformation dans laquelle il est perpendiculaire à ce plan. Or Z peut être «emprisonné» entre X et Y, soit au-dessus de ce plan, soit au-dessous, et les deux figures ainsi obtenues sont inverses optiques. Il s’ensuit que le composé se présente sous deux formes énantiomères, cela en l’absence de tout carbone asymétrique ou crypto-asymétrique: c’est l’atropo-isomérie, isomérie optique résultant d’une entrave à la libre rotation.Une autre conséquence curieuse de l’atropo-isomérie est que le composé (46) existe sous trois formes seulement: celles dans lesquelles les deux carbones marqués d’un astérisque ont même configuration, soit un composé dextrogyre et son inverse optique; celle dans laquelle ces carbones ont des configurations inverses. Ce dernier est un inactif indédoublable, bien qu’il soit impossible de lui imposer une conformation symétrique ou superposable à son image; mais, par des rotations judicieuses autour des liaisons CXYZ 漣noyau, la molécule se transforme spontanément en son image, seule condition nécessaire pour l’inactivité optique et l’unicité d’un tel lot.Stéréochimie des hétéroatomesLes orbitales de O, S, N, ont, comme pour le carbone, une distribution tétraédrique, mais, tant que ces éléments sont diou trivalents, le tétraèdre ne peut être régulier; il le serait dans l’ion +(R)4 dans lequel l’azote est tétracoordonné.Il s’ensuit que l’oxygène ou le soufre ne peuvent constituer un site de chiralité tant qu’ils restent bivalents: R 漣O 漣R et R 漣S 漣R (R et R non asymétriques) n’existent que sous une forme inactive.Il n’en est plus, en principe, de même pour les ions oxonium R R Rt+ et les ions sulfonium R R RtS+ en admettant que l’orbitale non liante joue le rôle stérique d’une liaison. Expérimentalement, on a constaté que les ions oxonium dissymétriques sont indédoublables en inverses optiques, alors que le même phénomène ne se présente pas pour le soufre, le sélénium, le tellure. Le problème est le même pour l’azote: une amine tertiaire dissymétrique s’équilibre rapidement avec son inverse optique (47) et on n’a pas réussi à dédoubler une amine tertiaire en inverses optiques, tout au moins en série acyclique ou monocyclique.En revanche, si l’orbitale non liante est utilisée dans une liaison avec un quatrième substituant, le dédoublement devient facile; tel est le cas des ions ammonium asymétriques +R1R2R3R4 et des aminoxydes ONR1R2R3.L’azote doublement lié est beaucoup moins sensible à cette conversion de configuration; c’est ainsi que les dérivés (48) et (49) existent sous ces deux formes, difficilement interconvertibles; on a donné à cette isomérie le nom d’isomérie syn-anti ; elle est très voisine de l’isomérie cis-trans éthylénique.2. Stéréochimie dynamiqueEmpêchement stériqueLes atomes sont impénétrables; chacun d’eux est entouré d’une région, dite sphère de Van der Waals , qui en interdit l’accès. Pour cette raison, on ne peut concevoir aucune structure, ni aucune conformation dans laquelle divers atomes sont trop proches les uns des autres; une première manifestation de cette interférence est l’explication de l’atropo-isomérie, et la prédominance de la position équatoriale des substituants volumineux. De même, l’accès à des molécules très encombrées (carbone uni à quatre substituants très ramifiés) est en général très difficile, parfois impossible.D’autre part, les substituants d’un carbone et les proches ramifications de ses voisins immédiats forment écran vis-à-vis des agents nucléophiles qui doivent attaquer ce carbone (processus SN2); c’est ainsi que les carbones primaires sont les plus accessibles, et qu’un halogénure tel que [(CH3)3C]3CBr ne peut subir aucune substitution de processus S2. On a donné à cet effet d’écran le nom d’empêchement stérique. La libre rotation atténue parfois l’empêchement stérique, car la gêne stérique peut disparaître chez certaines conformations; c’est pourquoi le phénomène est beaucoup plus net dans le cas de molécules rigides; l’empêchement stérique des radicaux R1 et R2 ralentit ou entrave de nombreuses réactions des composés (50). Mais l’effet stérique n’est pas toujours une diminution de la réactivité: dans le bromure [(CH3)3C]3CBr, les trois radicaux tertiobutyle interfèrent et s’écartent; on dit qu’il existe une compression stérique; celle-ci diminue lors d’un départ éventuel de l’ion Br- qui libère le carbocation [(CH3)3C]3C+ dans lequel C+ et les trois carbones qui lui sont directement reliés sont coplanaires. En conséquence, ce carbocation se forme très facilement et les réactions dites S1, dans lesquelles ce carbocation intervient, deviennent d’une étonnante facilité: on a donné à ce phénomène le nom d’accélération stérique.Enfin, l’empêchement stérique peut avoir d’autres conséquences; des radicaux encombrants entravent la coplanéité de deux noyaux benzéniques directement reliés; il s’ensuit une diminution de la conjugaison qui peut influer dans un sens ou dans l’autre sur la réactivité de la molécule: alors que le butadiène n’a qu’une désaturation moyenne, un dérivé substitué par des radicaux R très encombrants (R)2C=C(R) 漣C(R)=CR2 se comporte parfois comme le biradical:
alors qu’il n’y en a que six: un cis inactif, un cis droit, un cis gauche, un trans inactif, un trans droit, un trans gauche; le cumul des deux conventions laisserait supposer que le trans inactif est un racémique, car sa projection ne peut présenter un axe de symétrie, mais, dans l’espace, une de ses conformations possède un centre de symétrie.Isomérie spiranniqueOn appelle spiranne une molécule possédant un seul carbone commun à deux cycles. Les plans moyens de ces cycles sont perpendiculaires; si les cycles sont pairs et si on introduit, dans chacun d’eux, un substituant sur le carbone diamétralement opposé au carbone commun, la formule (41) ne comporte aucun carbone asymétrique, sensu stricto .Mais les vecteurs HXet HYsont perpendiculaires dans l’espace; ils peuvent être soit dextrorsomes, soit sinistrorsomes, et les deux configurations sont différentes et non superposables mais inverses optiques: en conclusion, à la formule (41) correspondent deux énantiomères.Le même raisonnement est valable pour les composés (42) et (43), qui sont effectivement dédoublables en inverses optiques.On appelle parfois carbone «crypto-asymétrique» le carbone commun aux deux cycles, ou le carbone commun à un cycle et à une double liaison, ou encore le carbone commun à deux doubles liaisons.Atropo-isomérieL’existence d’un seul carbone asymétrique ou cryptoasymétrique dans une molécule est une condition suffisante à l’existence des énantiomères; on a cru longtemps qu’elle était nécessaire; il n’en est rien.Le noyau benzénique est plan et les six liaisons qui en émanent sont dans ce plan et forment, deux à deux, des angles de 600.Le diphényle est l’hydrocarbure (44). Il est sensiblement plan, la mésomérie se transmettant d’un noyau à l’autre et donnant une grande stabilité à la structure dans laquelle les deux noyaux sont coplanaires. Cependant, cette planéité n’est pas rigoureuse, les hydrogènes explicités se gênant légèrement; il n’en est plus de même si nous substituons à ces hydrogènes des radicaux volumineux; dans un tel composé, les plans de deux noyaux forment un angle notable, et les molécules s’écartent peu de la position dans laquelle ils sont perpendiculaires.Soit maintenant un dérivé (45) trisubstitué, avec X et Y différents. L’encombrement dû à X, Y, Z entrave complètement la libre rotation autour de la liaison qui unit les noyaux. Il s’ensuit que si nous supposons le noyau de gauche dans le plan de figure, le noyau de droite oscille peu de part et d’autre de la conformation dans laquelle il est perpendiculaire à ce plan. Or Z peut être «emprisonné» entre X et Y, soit au-dessus de ce plan, soit au-dessous, et les deux figures ainsi obtenues sont inverses optiques. Il s’ensuit que le composé se présente sous deux formes énantiomères, cela en l’absence de tout carbone asymétrique ou crypto-asymétrique: c’est l’atropo-isomérie, isomérie optique résultant d’une entrave à la libre rotation.Une autre conséquence curieuse de l’atropo-isomérie est que le composé (46) existe sous trois formes seulement: celles dans lesquelles les deux carbones marqués d’un astérisque ont même configuration, soit un composé dextrogyre et son inverse optique; celle dans laquelle ces carbones ont des configurations inverses. Ce dernier est un inactif indédoublable, bien qu’il soit impossible de lui imposer une conformation symétrique ou superposable à son image; mais, par des rotations judicieuses autour des liaisons CXYZ 漣noyau, la molécule se transforme spontanément en son image, seule condition nécessaire pour l’inactivité optique et l’unicité d’un tel lot.Stéréochimie des hétéroatomesLes orbitales de O, S, N, ont, comme pour le carbone, une distribution tétraédrique, mais, tant que ces éléments sont diou trivalents, le tétraèdre ne peut être régulier; il le serait dans l’ion +(R)4 dans lequel l’azote est tétracoordonné.Il s’ensuit que l’oxygène ou le soufre ne peuvent constituer un site de chiralité tant qu’ils restent bivalents: R 漣O 漣R et R 漣S 漣R (R et R non asymétriques) n’existent que sous une forme inactive.Il n’en est plus, en principe, de même pour les ions oxonium R R Rt+ et les ions sulfonium R R RtS+ en admettant que l’orbitale non liante joue le rôle stérique d’une liaison. Expérimentalement, on a constaté que les ions oxonium dissymétriques sont indédoublables en inverses optiques, alors que le même phénomène ne se présente pas pour le soufre, le sélénium, le tellure. Le problème est le même pour l’azote: une amine tertiaire dissymétrique s’équilibre rapidement avec son inverse optique (47) et on n’a pas réussi à dédoubler une amine tertiaire en inverses optiques, tout au moins en série acyclique ou monocyclique.En revanche, si l’orbitale non liante est utilisée dans une liaison avec un quatrième substituant, le dédoublement devient facile; tel est le cas des ions ammonium asymétriques +R1R2R3R4 et des aminoxydes ONR1R2R3.L’azote doublement lié est beaucoup moins sensible à cette conversion de configuration; c’est ainsi que les dérivés (48) et (49) existent sous ces deux formes, difficilement interconvertibles; on a donné à cette isomérie le nom d’isomérie syn-anti ; elle est très voisine de l’isomérie cis-trans éthylénique.2. Stéréochimie dynamiqueEmpêchement stériqueLes atomes sont impénétrables; chacun d’eux est entouré d’une région, dite sphère de Van der Waals , qui en interdit l’accès. Pour cette raison, on ne peut concevoir aucune structure, ni aucune conformation dans laquelle divers atomes sont trop proches les uns des autres; une première manifestation de cette interférence est l’explication de l’atropo-isomérie, et la prédominance de la position équatoriale des substituants volumineux. De même, l’accès à des molécules très encombrées (carbone uni à quatre substituants très ramifiés) est en général très difficile, parfois impossible.D’autre part, les substituants d’un carbone et les proches ramifications de ses voisins immédiats forment écran vis-à-vis des agents nucléophiles qui doivent attaquer ce carbone (processus SN2); c’est ainsi que les carbones primaires sont les plus accessibles, et qu’un halogénure tel que [(CH3)3C]3CBr ne peut subir aucune substitution de processus S2. On a donné à cet effet d’écran le nom d’empêchement stérique. La libre rotation atténue parfois l’empêchement stérique, car la gêne stérique peut disparaître chez certaines conformations; c’est pourquoi le phénomène est beaucoup plus net dans le cas de molécules rigides; l’empêchement stérique des radicaux R1 et R2 ralentit ou entrave de nombreuses réactions des composés (50). Mais l’effet stérique n’est pas toujours une diminution de la réactivité: dans le bromure [(CH3)3C]3CBr, les trois radicaux tertiobutyle interfèrent et s’écartent; on dit qu’il existe une compression stérique; celle-ci diminue lors d’un départ éventuel de l’ion Br- qui libère le carbocation [(CH3)3C]3C+ dans lequel C+ et les trois carbones qui lui sont directement reliés sont coplanaires. En conséquence, ce carbocation se forme très facilement et les réactions dites S1, dans lesquelles ce carbocation intervient, deviennent d’une étonnante facilité: on a donné à ce phénomène le nom d’accélération stérique.Enfin, l’empêchement stérique peut avoir d’autres conséquences; des radicaux encombrants entravent la coplanéité de deux noyaux benzéniques directement reliés; il s’ensuit une diminution de la conjugaison qui peut influer dans un sens ou dans l’autre sur la réactivité de la molécule: alors que le butadiène n’a qu’une désaturation moyenne, un dérivé substitué par des radicaux R très encombrants (R)2C=C(R) 漣C(R)=CR2 se comporte parfois comme le biradical: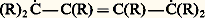 l’encombrement s’opposant à une disposition coplanaire des doubles liaisons favorisant la conjugaison.Fermeture des chaînesLe passage d’un composé acyclique à un composé cyclique dépend de deux facteurs: la probabilité de position favorable, qui décroît lorsque le nombre n d’atomes du cycle escompté augmente, et la tension qui diminue lorsque ce nombre croît de 2 à 5, pour devenir nulle ensuite. De l’antagonisme de ces facteurs découle une grande irrégularité dans les «facilités de cyclisation». Laissant de côté le cycle à deux atomes (double liaison) qui est un peu particulier, on peut dire que la facilité de fermeture (nombre de réactions cyclisantes connues), en fonction de n , se classe dans l’ordre décroissant suivant: 5, 6, 7, 3, n 礪 7 et 4. Pendant très longtemps, on n’avait connu qu’une réaction de cyclisation quadrangulaire; cet ordre est sujet à modifications du fait de la découverte de nouvelles réactions.Inversion de WaldenSoit (51) la projection conventionnelle d’un composé actif (d ). Lorsqu’on effectue sur lui une réaction réelle remplaçant, dans la formule plane, T par T , quelle sera la projection conventionnelle de T , (52) ou (53)? L’expérience montre qu’on ne peut rien conclure en général, dans l’ignorance du processus réactionnel; en effet, d peut conduire à d (rétention de structure), à l (inversion de Walden), au racémique d + l (racémisation totale), à un mélange de d et l soit avec rétention prépondérante, soit avec inversion prépondérante (racémisation partielle).L’inversion de Walden se symbolise: d 0l .On appelle taux de déracémisation le quotient du pouvoir rotatoire du mélange (d + l ) par celui de l (ou d ) pur. On lui donne un signe: + 1 dans le cas de la rétention, 漣 1 dans le cas de l’inversion, zéro dans le cas de la racémisation totale, positif en cas de rétention prépondérante, négatif dans le cas contraire.Il est à remarquer que d et d possèdent ou non des pouvoirs rotatoires de même signe, et que la connaissance des configurations absolues, ou tout au moins relatives, est indispensable pour fixer le signe des taux de déracémisation différents de zéro; cette remarque ne simplifie pas le problème.On peut citer quelques règles:a ) il y a toujours rétention de structure si la substitution n’affecte pas une liaison directe du carbone;b ) les réactions de type S2 pur se font avec inversion totale;c ) les réactions de type S1 pur se font avec racémisation totale;d ) la racémisation partielle, avec inversion prépondérante, s’observe quand le processus est intermédiaire entre S2 et S1 (mésomécanismes, attaques nucléophile et électrophile concertées);e ) quelques réactions S1 conduisent cependant à la rétention totale.ÉpimérisationSi un groupe 漣CHOH 漣 est voisin d’un groupe C-2, son attaque par l’ion OH- se fait avec inversion de Walden. Si R est symétrique, la réaction (54) étant réversible, on arrive, par raison de symétrie, au mélange racémique (d + l ).Mais si R renferme d’autres carbones asymétriques, l’inversion n’intéresse que celui des carbones hydroxylés voisin du groupe C-2. À l’équilibre, les deux ions gluconate et mannonate (55) apparaissent en quantités inégales du fait que le reste de la molécule n’est plus symétrique.On réalise ainsi le passage partiel de l’acide gluconique à l’acide mannonique. Cette transformation, qui fait passer partiellement d’un diastéréo-isomère à un autre, porte le nom de stéréomutation ou d’épimérisation: l’acide gluconique et l’acide mannonique sont des épimères. L’épimérisation a joué un rôle fondamental dans l’étude des oses.Additions aux liaisons multiplesA priori, de l’addition de X2 sur un alcyne , on peut prévoir deux isomères: un dérivé alcénique cis et un autre trans. Si X est un halogène, le dérivé trans domine, sans toutefois être exclusif. Pour X = H, le résultat va dépendre du mode d’hydrogénation ; l’hydrogénation chimique: (Na + H2O) conduit surtout à l’oléfine trans ; l’hydrogénation catalytique (Pd colloïdal) engendre l’oléfine cis avec d’excellents rendements.Les mêmes problèmes se posent pour une oléfine R 漣CH=CH 漣R ; celle-ci peut avoir la configuration cis ou la configuration trans ; l’addition de X2 conduit, a priori, à l’un des isomères (56) et (57), généralement racémiques.Le passage de l’oléfine cis à un dérivé érythro s’appelle une cis -addition; son passage à un dérivé thréo s’appelle une trans -addition; réciproquement, le passage de l’oléfine trans à l’isomère thréo est une cis -addition; le passage à l’isomère érythro est une trans -addition.Dédoublement des racémiquesDédoublement spontanéEn principe, deux inverses optiques ayant mêmes constantes physiques scalaires sont indédoublables par les procédés de l’analyse immédiate. Toutefois, la cristallisation fractionnée n’est pas toujours un phénomène d’équilibre parfait (on observe fréquemment des sursaturations).Lorsque la solution sursaturée d’un racémique commence à cristalliser, il est possible qu’un seul des inverses optiques se dépose en un point et que son inverse optique le fasse à quelque distance de là, de sorte qu’il peut apparaître, ou simultanément ou successivement, les deux sortes de cristaux.Ceux-ci sont classés en deux lots, soit par analyse polarimétrique, ce qui est pratiquement inextricable, soit d’après une hémiédrie extérieurement apparente. Ces conditions sont très exigeantes et rarement réalisées. En effet, de nombreux inverses optiques s’associent pour engendrer des cristaux uniques de composition (d + l ); on dit qu’on a affaire à un racémique vrai. C’est alors lui qui se dépose au cours de la cristallisation.Ce n’est que lorsqu’il n’existe pas de racémique vrai que l’on peut espérer un dédoublement spontané. Mais il faut encore que les cristaux formés soient assez gros pour être séparés (au besoin sous le microscope), et que les formes d et l se distinguent extérieurement.D’ailleurs, les cristaux recueillis sont souillés de liqueur mère et quelques erreurs de classement sont possibles; il convient donc de recristalliser les deux lots. Si la composition d’un lot n’est pas très différente de l’un des antipodes purs, une seule recristallisation fractionnée livre ce dernier à l’état pur.Il est arrivé parfois que la solution sursaturée d’un racémique ne laisse déposer qu’un des inverses optiques; il s’agit probablement de l’amorçage unilatéral de la cristallisation par un germe isomorphe apporté par l’atmosphère; il ne faut pas trop compter sur cette réussite aléatoire. En fait, on a rencontré seulement quelques dizaines de dédoublements spontanés.Si les deux inverses optiques se combinent en un racémique vrai, il existe encore la ressource de les transformer en deux dérivés ne présentant plus cette particularité; c’est ainsi que les acides tartriques actifs (D) et (L) formant un racémique vrai sont spontanément inséparables, mais Pasteur, les ayant transformés en tartrates mixtes de sodium et d’ammonium qui ne s’associent plus, a pu isoler les sels D et L sans trop de difficultés.Méthodes chimiques (recours aux diastéréo-isomères)Soit, d’une part, un mélange racémique d + l et, d’autre part, un composé 炙 actif sur la lumière polarisée, choisi de façon à se combiner à la fois à d et à l . Il en résulte un mélange de deux composés que nous symboliserons par d 炙 et l 炙. Ces deux composés ne sont pas inverses optiques, car l’inverse optique de d 炙 est l ( étant l’inverse optique de 炙). En conséquence, d 炙 et l 炙 sont des diastéréo-isomères présentant des propriétés physiques (en particulier des solubilités) différentes et, en principe tout au moins, séparables par cristallisation fractionnée.La séparation effectuée, on régénère d et l , à partir de d 炙 et l 炙, par la réaction inverse de celle qui leur avait donné naissance. C’est particulièrement facile si d et l sont des acides et 炙 une amine (alcaloïde actif), ou réciproquement. C’est également possible si d et l sont des alcools et 炙 un acide actif, mais, lors de l’hydrolyse de l’ester formé, une racémisation est parfois à craindre. On peut d’ailleurs utiliser un intermédiaire: un alcool racémique est transformé (58), par l’anhydride phtalique, en phtalate acide. Celui-ci forme, grâce au groupe 漣COOH, des sels avec les alcaloïdes actifs.Si le dédoublement échoue dans l’emploi de 炙, du fait que d 炙 et l 炙 ont des solubilités trop proches, il ne faut pas se décourager, mais faire appel à d’autres composés actifs auxiliaires 炙 , 炙 , etc.Cette technique est utilisée dans la préparation de produits pharmaceutiques synthétiques, obtenus souvent sous forme racémique, et dont seule une variété optique est physiologiquement active, l’autre pouvant même être toxique.Méthodes biologiquesEn principe l et d agissent sur 炙 avec des vitesses différentes, et il n’est pas interdit d’espérer que l’action de 炙 en défaut sur le racémique d + l laissera un résidu enrichi relativement en d ou en l ; cependant, tant que d , l , 炙 sont simples, les différences de vitesse ne permettent qu’un enrichissement insignifiant.Il n’en est plus de même si 炙 est une enzyme. Ces composés sont extrêmement stéréospécifiques et peuvent, par exemple, transformer l (nous dirons simplement que l est détruit), en laissant d inattaqué; c’est ainsi que Penicillum glaucum , la moisissure du pain, sécrète une enzyme capable de détruire la forme lévogyre de l’alcool racémique:
l’encombrement s’opposant à une disposition coplanaire des doubles liaisons favorisant la conjugaison.Fermeture des chaînesLe passage d’un composé acyclique à un composé cyclique dépend de deux facteurs: la probabilité de position favorable, qui décroît lorsque le nombre n d’atomes du cycle escompté augmente, et la tension qui diminue lorsque ce nombre croît de 2 à 5, pour devenir nulle ensuite. De l’antagonisme de ces facteurs découle une grande irrégularité dans les «facilités de cyclisation». Laissant de côté le cycle à deux atomes (double liaison) qui est un peu particulier, on peut dire que la facilité de fermeture (nombre de réactions cyclisantes connues), en fonction de n , se classe dans l’ordre décroissant suivant: 5, 6, 7, 3, n 礪 7 et 4. Pendant très longtemps, on n’avait connu qu’une réaction de cyclisation quadrangulaire; cet ordre est sujet à modifications du fait de la découverte de nouvelles réactions.Inversion de WaldenSoit (51) la projection conventionnelle d’un composé actif (d ). Lorsqu’on effectue sur lui une réaction réelle remplaçant, dans la formule plane, T par T , quelle sera la projection conventionnelle de T , (52) ou (53)? L’expérience montre qu’on ne peut rien conclure en général, dans l’ignorance du processus réactionnel; en effet, d peut conduire à d (rétention de structure), à l (inversion de Walden), au racémique d + l (racémisation totale), à un mélange de d et l soit avec rétention prépondérante, soit avec inversion prépondérante (racémisation partielle).L’inversion de Walden se symbolise: d 0l .On appelle taux de déracémisation le quotient du pouvoir rotatoire du mélange (d + l ) par celui de l (ou d ) pur. On lui donne un signe: + 1 dans le cas de la rétention, 漣 1 dans le cas de l’inversion, zéro dans le cas de la racémisation totale, positif en cas de rétention prépondérante, négatif dans le cas contraire.Il est à remarquer que d et d possèdent ou non des pouvoirs rotatoires de même signe, et que la connaissance des configurations absolues, ou tout au moins relatives, est indispensable pour fixer le signe des taux de déracémisation différents de zéro; cette remarque ne simplifie pas le problème.On peut citer quelques règles:a ) il y a toujours rétention de structure si la substitution n’affecte pas une liaison directe du carbone;b ) les réactions de type S2 pur se font avec inversion totale;c ) les réactions de type S1 pur se font avec racémisation totale;d ) la racémisation partielle, avec inversion prépondérante, s’observe quand le processus est intermédiaire entre S2 et S1 (mésomécanismes, attaques nucléophile et électrophile concertées);e ) quelques réactions S1 conduisent cependant à la rétention totale.ÉpimérisationSi un groupe 漣CHOH 漣 est voisin d’un groupe C-2, son attaque par l’ion OH- se fait avec inversion de Walden. Si R est symétrique, la réaction (54) étant réversible, on arrive, par raison de symétrie, au mélange racémique (d + l ).Mais si R renferme d’autres carbones asymétriques, l’inversion n’intéresse que celui des carbones hydroxylés voisin du groupe C-2. À l’équilibre, les deux ions gluconate et mannonate (55) apparaissent en quantités inégales du fait que le reste de la molécule n’est plus symétrique.On réalise ainsi le passage partiel de l’acide gluconique à l’acide mannonique. Cette transformation, qui fait passer partiellement d’un diastéréo-isomère à un autre, porte le nom de stéréomutation ou d’épimérisation: l’acide gluconique et l’acide mannonique sont des épimères. L’épimérisation a joué un rôle fondamental dans l’étude des oses.Additions aux liaisons multiplesA priori, de l’addition de X2 sur un alcyne , on peut prévoir deux isomères: un dérivé alcénique cis et un autre trans. Si X est un halogène, le dérivé trans domine, sans toutefois être exclusif. Pour X = H, le résultat va dépendre du mode d’hydrogénation ; l’hydrogénation chimique: (Na + H2O) conduit surtout à l’oléfine trans ; l’hydrogénation catalytique (Pd colloïdal) engendre l’oléfine cis avec d’excellents rendements.Les mêmes problèmes se posent pour une oléfine R 漣CH=CH 漣R ; celle-ci peut avoir la configuration cis ou la configuration trans ; l’addition de X2 conduit, a priori, à l’un des isomères (56) et (57), généralement racémiques.Le passage de l’oléfine cis à un dérivé érythro s’appelle une cis -addition; son passage à un dérivé thréo s’appelle une trans -addition; réciproquement, le passage de l’oléfine trans à l’isomère thréo est une cis -addition; le passage à l’isomère érythro est une trans -addition.Dédoublement des racémiquesDédoublement spontanéEn principe, deux inverses optiques ayant mêmes constantes physiques scalaires sont indédoublables par les procédés de l’analyse immédiate. Toutefois, la cristallisation fractionnée n’est pas toujours un phénomène d’équilibre parfait (on observe fréquemment des sursaturations).Lorsque la solution sursaturée d’un racémique commence à cristalliser, il est possible qu’un seul des inverses optiques se dépose en un point et que son inverse optique le fasse à quelque distance de là, de sorte qu’il peut apparaître, ou simultanément ou successivement, les deux sortes de cristaux.Ceux-ci sont classés en deux lots, soit par analyse polarimétrique, ce qui est pratiquement inextricable, soit d’après une hémiédrie extérieurement apparente. Ces conditions sont très exigeantes et rarement réalisées. En effet, de nombreux inverses optiques s’associent pour engendrer des cristaux uniques de composition (d + l ); on dit qu’on a affaire à un racémique vrai. C’est alors lui qui se dépose au cours de la cristallisation.Ce n’est que lorsqu’il n’existe pas de racémique vrai que l’on peut espérer un dédoublement spontané. Mais il faut encore que les cristaux formés soient assez gros pour être séparés (au besoin sous le microscope), et que les formes d et l se distinguent extérieurement.D’ailleurs, les cristaux recueillis sont souillés de liqueur mère et quelques erreurs de classement sont possibles; il convient donc de recristalliser les deux lots. Si la composition d’un lot n’est pas très différente de l’un des antipodes purs, une seule recristallisation fractionnée livre ce dernier à l’état pur.Il est arrivé parfois que la solution sursaturée d’un racémique ne laisse déposer qu’un des inverses optiques; il s’agit probablement de l’amorçage unilatéral de la cristallisation par un germe isomorphe apporté par l’atmosphère; il ne faut pas trop compter sur cette réussite aléatoire. En fait, on a rencontré seulement quelques dizaines de dédoublements spontanés.Si les deux inverses optiques se combinent en un racémique vrai, il existe encore la ressource de les transformer en deux dérivés ne présentant plus cette particularité; c’est ainsi que les acides tartriques actifs (D) et (L) formant un racémique vrai sont spontanément inséparables, mais Pasteur, les ayant transformés en tartrates mixtes de sodium et d’ammonium qui ne s’associent plus, a pu isoler les sels D et L sans trop de difficultés.Méthodes chimiques (recours aux diastéréo-isomères)Soit, d’une part, un mélange racémique d + l et, d’autre part, un composé 炙 actif sur la lumière polarisée, choisi de façon à se combiner à la fois à d et à l . Il en résulte un mélange de deux composés que nous symboliserons par d 炙 et l 炙. Ces deux composés ne sont pas inverses optiques, car l’inverse optique de d 炙 est l ( étant l’inverse optique de 炙). En conséquence, d 炙 et l 炙 sont des diastéréo-isomères présentant des propriétés physiques (en particulier des solubilités) différentes et, en principe tout au moins, séparables par cristallisation fractionnée.La séparation effectuée, on régénère d et l , à partir de d 炙 et l 炙, par la réaction inverse de celle qui leur avait donné naissance. C’est particulièrement facile si d et l sont des acides et 炙 une amine (alcaloïde actif), ou réciproquement. C’est également possible si d et l sont des alcools et 炙 un acide actif, mais, lors de l’hydrolyse de l’ester formé, une racémisation est parfois à craindre. On peut d’ailleurs utiliser un intermédiaire: un alcool racémique est transformé (58), par l’anhydride phtalique, en phtalate acide. Celui-ci forme, grâce au groupe 漣COOH, des sels avec les alcaloïdes actifs.Si le dédoublement échoue dans l’emploi de 炙, du fait que d 炙 et l 炙 ont des solubilités trop proches, il ne faut pas se décourager, mais faire appel à d’autres composés actifs auxiliaires 炙 , 炙 , etc.Cette technique est utilisée dans la préparation de produits pharmaceutiques synthétiques, obtenus souvent sous forme racémique, et dont seule une variété optique est physiologiquement active, l’autre pouvant même être toxique.Méthodes biologiquesEn principe l et d agissent sur 炙 avec des vitesses différentes, et il n’est pas interdit d’espérer que l’action de 炙 en défaut sur le racémique d + l laissera un résidu enrichi relativement en d ou en l ; cependant, tant que d , l , 炙 sont simples, les différences de vitesse ne permettent qu’un enrichissement insignifiant.Il n’en est plus de même si 炙 est une enzyme. Ces composés sont extrêmement stéréospécifiques et peuvent, par exemple, transformer l (nous dirons simplement que l est détruit), en laissant d inattaqué; c’est ainsi que Penicillum glaucum , la moisissure du pain, sécrète une enzyme capable de détruire la forme lévogyre de l’alcool racémique: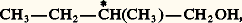 en laissant intacte la forme dextrogyre.L’alcool naturel (alcool amylique actif) est la forme lévogyre; une longue ébullition avec la soude le racémise. Par ce procédé, Le Bel a obtenu, avec un rendement qui ne saurait dépasser 50 p. 100, l’alcool dextrogyre à partir du produit naturel.L’action enzymatique est toujours aléatoire; elle ne peut être efficace que si (d + l ) n’est pas trop insoluble dans l’eau, seul milieu biologique. On peut toujours la tenter si l’on sait qu’une enzyme détruit un produit naturel; il est probable qu’elle laissera intact son inverse optique.Autres méthodesDeux inverses optiques ont, en principe, des solubilités différentes dans un solvant optiquement actif, mais les différences sont trop faibles pour rendre le procédé de dédoublement pratique; il en est de même de l’élution par un solvant actif d’un racémique fixé sur support inerte (Al23).Des résultats un peu meilleurs ont été obtenus par chromatographie sur support actif (D-lactose).Enfin, certains composés comme l’urée forment des clathrates cristallisés, sorte de moules bâtis autour d’une molécule; si l’on réalise ce clathrate autour d’une molécule active, il est dissymétrique. Mis au contact d’un racémique, seul l’un des inverses optiques peut en déplacer le composé actif emprisonné, à supposer qu’il n’en diffère pas trop par son volume et par sa forme; en détruisant le nouveau clathrate, on isole l’un des inverses optiques.Néanmoins, aucune de ces méthodes ne soutient la comparaison avec les trois premières, imaginées par Pasteur il y a plus d’un siècle. Toutes utilisent un auxiliaire dissymétrique; c’est évident, sauf peut-être pour la première où l’auxiliaire dissymétrique est l’opérateur qui sait distinguer sa droite de sa gauche, et pourrait, en théorie, être remplacé par une machine dissymétrique.Configurations absolues et configurations relativesToute la stéréochimie des sucres repose sur celle de l’aldéhyde D-glycérique (aldéhyde glycérique dextrogyre) qui pouvait a priori correspondre aux formules (59) et (60).On a choisi arbitrairement la première structure, et c’est bien longtemps plus tard qu’on a pu démontrer qu’elle était exacte.Les rayons X, dans leur emploi courant, ne peuvent pas aider à trancher entre les deux formules, et c’est grâce à des artifices que nous ne détaillerons pas qu’on a pu préciser la structure de l’aldéhyde D-glycérique et celle de quelques autres composés actifs.Par contre, il existe des méthodes permettant de déterminer les configurations relatives. Soit CXYZT, un composé donnant un racémique vrai, et supposons que l’un de ses inverses optiques se combine avec l’un des inverses optiques de CXYZT , et non avec l’autre; il est alors probable que le composé d’addition est CXYZT (d ) + CXYZT (l ); on l’appelle racémique actif , car il a en solution un pouvoir rotatoire égal à la somme algébrique de ceux de CXYZT (d ) et de CXYZT (l ). Un racémique actif est donc formé de deux composés actifs de configurations inverses; de celle de l’un, on déduit celle de l’autre et, si on connaît la configuration absolue de l’un d’eux, celle de l’autre en découle. Malheureusement, la formation d’un racémique actif suppose que T et T ont des encombrements très voisins, par exemple T = OH, T = NH2; aussi, la méthode des racémiques actifs est-elle d’un emploi très limité.Synthèse asymétriqueEn dehors de très rares dédoublements spontanés, l’obtention d’un corps optiquement actif suppose, quelque part, l’utilisation d’un autre composé actif. Le principe de Pasteur nous dit en effet qu’un composé actif ne peut résulter de l’action de réactifs inactifs. Ce principe paraît en contradiction avec l’asymétrie des composés biologiques et, comme toute biosynthèse fait appel au gaz carbonique, à l’eau, à l’azote libre, nitrique ou ammoniacal, on explique mal que l’essence de térébenthine, par exemple, possède un fort pouvoir rotatoire.On avait tenté d’expliquer l’asymétrie du monde vivant soit par l’action simultanée du champ magnétique terrestre et des courants telluriques, ce qui crée une dissymétrie du milieu, soit par la destruction sélective d’un des antipodes optiques par la lumière solaire qui est très légèrement polarisée circulairement. Des expériences dans lequelles un carbone asymétrique prend naissance dans les champs électriques et magnétiques les plus puissants n’ont jamais donné la moindre trace de composé actif, et les lumières polarisées circulairement les plus intenses n’ont conduit, et dans des cas très particuliers, qu’à des taux de déracémisation inférieurs à 1 p. 100. La synthèse asymétrique, dite de pemière espèce, reste à faire.Synthèse asymétrique de deuxième espècePrenons un exemple théorique. L’acide acrylique, CH2=CH 漣C2H, fixe le brome en conduisant à l’acide 見- 廓-dibromopropionique, CH2Br 漣CHBr 漣C2H, mais le produit ainsi obtenu est rigoureusement racémique. Si, par contre, on fixe le brome sur l’ester acrylique d’un alcool actif CH2=CH 漣COOR, on constate que l’acide dibromopropionique obtenu après hydrolyse n’est plus racémique; toutefois le taux de déracémisation n’atteint pas 1 p. 100. L’alcool actif étant récupérable peut, théoriquement, rentrer dans le cycle, et une quantité limitée de cet alcool conduirait à une quantité illimitée de dibromure actif. Le faible rendement de cette synthèse asymétrique s’explique par l’éloignement du carbone asymétrique inducteur (de l’alcool) et du carbone asymétrique induit (celui du groupe CH de l’ester). Mais, le principe admis, rien n’empêche de concevoir d’autres synthèses conduisant à des taux de déracémisation meilleurs; de fait, on en a observé qui atteignent 10 p. 100 et même 20 p. 100, et une expérience de même type, où malheureusement l’inducteur actif n’était pas intégralement récupérable, a conduit à un taux de 60 p. 100.Grâce à la grande spécificité des enzymes, on peut espérer d’elles un taux de déracémisation de 100 p. 100, et cette hypothèse permet, dans une certaine mesure, de rendre compte de l’asymétrie de la biosynthèse actuelle.Il faut faire un pas de plus pour expliquer l’origine de l’asymétrie du monde vivant; les composés qui président à la biosynthèse ont la propriété de se reproduire à leur propre image, mais cela suppose déjà une complication extrême.Sous l’influence des rayons solaires, l’eau, le gaz carbonique, l’azote engendrent de nombreux composés parmi lesquels des acides aminés; ceux-ci sont les éléments de base des enzymes et des autres facteurs de la biosynthèse.Les plus simples n’ont pu acquérir cette remarquable activité autocatalytique qu’en faisant appel à des centaines, peut-être des milliers d’acides aminés identiques ou différents; cela fait pressentir un nombre de diastéréo-isomères supérieur à 10100 (!) et, parmi eux, un très petit nombre, peut-être un seul d’entre eux, possédait cette activité.Or, la probabilité pour qu’au même instant apparaissent simultanément cette première protéine et son inverse optique est négligeable. Si nous la désignons par d , elle n’a pu créer à son image que des analogues d , eux-mêmes capables d’en engendrer de nouveaux, et cette première molécule active unique d a pu accaparer à son profit tous les éléments alors disponibles; le monde vivant est bâti sur le modèle d .Il n’y a aucun espoir de voir naître un jour son inverse optique sur la Terre; peut-être est-il un peu moins invraisemblable de le voir apparaître en un autre point de l’univers. Par contre, il n’est pas impossible que d’autres macromolécules organiques soient également dotées d’activité autocatalytique; elles engendreraient un monde vivant tout à fait différent de celui que nous connaissons, mais les biologistes estiment que cette probabilité n’excède pas 1/2 dans une sphère dont le rayon est de 104 années-lumière.Synthèse asymétrique de troisième espèce ou orientation stériqueDans la synthèse asymétrique de seconde espèce, la molécule active auxiliaire peut être séparée du produit synthétisé, et nous avons vu que les taux de déracémisation sont, en général, très faibles. Par contre, si l’on crée un carbone asymétrique à côté d’un autre, la configuration de ce dernier peut imposer au carbone asymétrique nouveau une configuration préférentielle, et parfois exclusive; le carbone asymétrique induit ne peut ici être séparé du carbone asymétrique inducteur sans rupture de l’édifice.C’est ainsi qu’en opposant à la cétone R 漣CHOH 漣CO 漣R l’organomagnésien R MgX, les deux diastéréo-isomères (61) et (62) prennent naissance en quantités inégales, et que parfois un seul d’entre eux se forme pratiquement. D’ailleurs, ces expériences sont parfois conduites à partir de composés racémiques. On aboutit alors à des racémiques diastéréo-isomères en quantités inégales. Elles peuvent même partir de composés inactifs indédoublables; par exemple, l’addition d’acide cyanhydrique au glyoxal:
en laissant intacte la forme dextrogyre.L’alcool naturel (alcool amylique actif) est la forme lévogyre; une longue ébullition avec la soude le racémise. Par ce procédé, Le Bel a obtenu, avec un rendement qui ne saurait dépasser 50 p. 100, l’alcool dextrogyre à partir du produit naturel.L’action enzymatique est toujours aléatoire; elle ne peut être efficace que si (d + l ) n’est pas trop insoluble dans l’eau, seul milieu biologique. On peut toujours la tenter si l’on sait qu’une enzyme détruit un produit naturel; il est probable qu’elle laissera intact son inverse optique.Autres méthodesDeux inverses optiques ont, en principe, des solubilités différentes dans un solvant optiquement actif, mais les différences sont trop faibles pour rendre le procédé de dédoublement pratique; il en est de même de l’élution par un solvant actif d’un racémique fixé sur support inerte (Al23).Des résultats un peu meilleurs ont été obtenus par chromatographie sur support actif (D-lactose).Enfin, certains composés comme l’urée forment des clathrates cristallisés, sorte de moules bâtis autour d’une molécule; si l’on réalise ce clathrate autour d’une molécule active, il est dissymétrique. Mis au contact d’un racémique, seul l’un des inverses optiques peut en déplacer le composé actif emprisonné, à supposer qu’il n’en diffère pas trop par son volume et par sa forme; en détruisant le nouveau clathrate, on isole l’un des inverses optiques.Néanmoins, aucune de ces méthodes ne soutient la comparaison avec les trois premières, imaginées par Pasteur il y a plus d’un siècle. Toutes utilisent un auxiliaire dissymétrique; c’est évident, sauf peut-être pour la première où l’auxiliaire dissymétrique est l’opérateur qui sait distinguer sa droite de sa gauche, et pourrait, en théorie, être remplacé par une machine dissymétrique.Configurations absolues et configurations relativesToute la stéréochimie des sucres repose sur celle de l’aldéhyde D-glycérique (aldéhyde glycérique dextrogyre) qui pouvait a priori correspondre aux formules (59) et (60).On a choisi arbitrairement la première structure, et c’est bien longtemps plus tard qu’on a pu démontrer qu’elle était exacte.Les rayons X, dans leur emploi courant, ne peuvent pas aider à trancher entre les deux formules, et c’est grâce à des artifices que nous ne détaillerons pas qu’on a pu préciser la structure de l’aldéhyde D-glycérique et celle de quelques autres composés actifs.Par contre, il existe des méthodes permettant de déterminer les configurations relatives. Soit CXYZT, un composé donnant un racémique vrai, et supposons que l’un de ses inverses optiques se combine avec l’un des inverses optiques de CXYZT , et non avec l’autre; il est alors probable que le composé d’addition est CXYZT (d ) + CXYZT (l ); on l’appelle racémique actif , car il a en solution un pouvoir rotatoire égal à la somme algébrique de ceux de CXYZT (d ) et de CXYZT (l ). Un racémique actif est donc formé de deux composés actifs de configurations inverses; de celle de l’un, on déduit celle de l’autre et, si on connaît la configuration absolue de l’un d’eux, celle de l’autre en découle. Malheureusement, la formation d’un racémique actif suppose que T et T ont des encombrements très voisins, par exemple T = OH, T = NH2; aussi, la méthode des racémiques actifs est-elle d’un emploi très limité.Synthèse asymétriqueEn dehors de très rares dédoublements spontanés, l’obtention d’un corps optiquement actif suppose, quelque part, l’utilisation d’un autre composé actif. Le principe de Pasteur nous dit en effet qu’un composé actif ne peut résulter de l’action de réactifs inactifs. Ce principe paraît en contradiction avec l’asymétrie des composés biologiques et, comme toute biosynthèse fait appel au gaz carbonique, à l’eau, à l’azote libre, nitrique ou ammoniacal, on explique mal que l’essence de térébenthine, par exemple, possède un fort pouvoir rotatoire.On avait tenté d’expliquer l’asymétrie du monde vivant soit par l’action simultanée du champ magnétique terrestre et des courants telluriques, ce qui crée une dissymétrie du milieu, soit par la destruction sélective d’un des antipodes optiques par la lumière solaire qui est très légèrement polarisée circulairement. Des expériences dans lequelles un carbone asymétrique prend naissance dans les champs électriques et magnétiques les plus puissants n’ont jamais donné la moindre trace de composé actif, et les lumières polarisées circulairement les plus intenses n’ont conduit, et dans des cas très particuliers, qu’à des taux de déracémisation inférieurs à 1 p. 100. La synthèse asymétrique, dite de pemière espèce, reste à faire.Synthèse asymétrique de deuxième espècePrenons un exemple théorique. L’acide acrylique, CH2=CH 漣C2H, fixe le brome en conduisant à l’acide 見- 廓-dibromopropionique, CH2Br 漣CHBr 漣C2H, mais le produit ainsi obtenu est rigoureusement racémique. Si, par contre, on fixe le brome sur l’ester acrylique d’un alcool actif CH2=CH 漣COOR, on constate que l’acide dibromopropionique obtenu après hydrolyse n’est plus racémique; toutefois le taux de déracémisation n’atteint pas 1 p. 100. L’alcool actif étant récupérable peut, théoriquement, rentrer dans le cycle, et une quantité limitée de cet alcool conduirait à une quantité illimitée de dibromure actif. Le faible rendement de cette synthèse asymétrique s’explique par l’éloignement du carbone asymétrique inducteur (de l’alcool) et du carbone asymétrique induit (celui du groupe CH de l’ester). Mais, le principe admis, rien n’empêche de concevoir d’autres synthèses conduisant à des taux de déracémisation meilleurs; de fait, on en a observé qui atteignent 10 p. 100 et même 20 p. 100, et une expérience de même type, où malheureusement l’inducteur actif n’était pas intégralement récupérable, a conduit à un taux de 60 p. 100.Grâce à la grande spécificité des enzymes, on peut espérer d’elles un taux de déracémisation de 100 p. 100, et cette hypothèse permet, dans une certaine mesure, de rendre compte de l’asymétrie de la biosynthèse actuelle.Il faut faire un pas de plus pour expliquer l’origine de l’asymétrie du monde vivant; les composés qui président à la biosynthèse ont la propriété de se reproduire à leur propre image, mais cela suppose déjà une complication extrême.Sous l’influence des rayons solaires, l’eau, le gaz carbonique, l’azote engendrent de nombreux composés parmi lesquels des acides aminés; ceux-ci sont les éléments de base des enzymes et des autres facteurs de la biosynthèse.Les plus simples n’ont pu acquérir cette remarquable activité autocatalytique qu’en faisant appel à des centaines, peut-être des milliers d’acides aminés identiques ou différents; cela fait pressentir un nombre de diastéréo-isomères supérieur à 10100 (!) et, parmi eux, un très petit nombre, peut-être un seul d’entre eux, possédait cette activité.Or, la probabilité pour qu’au même instant apparaissent simultanément cette première protéine et son inverse optique est négligeable. Si nous la désignons par d , elle n’a pu créer à son image que des analogues d , eux-mêmes capables d’en engendrer de nouveaux, et cette première molécule active unique d a pu accaparer à son profit tous les éléments alors disponibles; le monde vivant est bâti sur le modèle d .Il n’y a aucun espoir de voir naître un jour son inverse optique sur la Terre; peut-être est-il un peu moins invraisemblable de le voir apparaître en un autre point de l’univers. Par contre, il n’est pas impossible que d’autres macromolécules organiques soient également dotées d’activité autocatalytique; elles engendreraient un monde vivant tout à fait différent de celui que nous connaissons, mais les biologistes estiment que cette probabilité n’excède pas 1/2 dans une sphère dont le rayon est de 104 années-lumière.Synthèse asymétrique de troisième espèce ou orientation stériqueDans la synthèse asymétrique de seconde espèce, la molécule active auxiliaire peut être séparée du produit synthétisé, et nous avons vu que les taux de déracémisation sont, en général, très faibles. Par contre, si l’on crée un carbone asymétrique à côté d’un autre, la configuration de ce dernier peut imposer au carbone asymétrique nouveau une configuration préférentielle, et parfois exclusive; le carbone asymétrique induit ne peut ici être séparé du carbone asymétrique inducteur sans rupture de l’édifice.C’est ainsi qu’en opposant à la cétone R 漣CHOH 漣CO 漣R l’organomagnésien R MgX, les deux diastéréo-isomères (61) et (62) prennent naissance en quantités inégales, et que parfois un seul d’entre eux se forme pratiquement. D’ailleurs, ces expériences sont parfois conduites à partir de composés racémiques. On aboutit alors à des racémiques diastéréo-isomères en quantités inégales. Elles peuvent même partir de composés inactifs indédoublables; par exemple, l’addition d’acide cyanhydrique au glyoxal: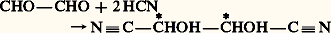 conduit à un mélange en quantités inégales du nitrile tartrique racémique et du nitrile tartrique méso; on considère que l’intermédiaire est le mononitrile racémique N 令C 漣CHOH 漣CHO, ce qui ramène au type précédemment exposé.3. Chiralité et synthèse asymétriqueDurant les dix dernières années, de grands progrès ont été accomplis dans les méthodes d’études des structures chirales. Celles-ci sont importantes car elles interviennent dans beaucoup de processus biologiques. La majorité des molécules d’intérêt biologique sont chirales (polypeptides, enzymes, stéroïdes, prostaglandines, etc.). Les plus simples d’entre elles peuvent être obtenues par synthèse totale, mais seul l’énantiomère ayant la configuration absolue désirée (cf. supra , fig. 1) doit être sélectivement préparé.Le terme chiralité est dérivé du nom grec kheir ( 﨑﨎晴福, main) et caractérise deux objets qui, comme une main droite et une main gauche, sont différents tout en paraissant similaires. La distinction ne réside pas dans des dimensions ou des formes respectives. Une main gauche et une main droite ne peuvent être amenées en superposition, elles se déduisent l’une de l’autre par une symétrie par rapport à un plan. Presque tous les constituants chimiques des systèmes vivants sont chiraux: polypeptides, protéines, glucides, acides nucléiques; la chiralité est une des caractéristiques des organismes vivants. C’est, toutefois, à une date relativement récente (1950-1960) que ce terme utilisé par le physicien lord Kelvin au début du siècle a été introduit en chimie par Cahn, Ingold, Prelog, Mislow. Il devenait, en effet, indispensable de disposer d’un mot caractérisant des espèces moléculaires les plus variées qui peuvent exister sous forme de deux stéréo-isomères, image l’un de l’autre par rapport à un plan miroir. Le mot chiralité ne fait pas appel à des considérations de symétrie, il s’applique à tout composé pouvant avoir une image non superposable, appelé composé antipode ou énantiomère . Par exemple, l’acide lactique (formule 63) est dépourvu d’éléments de symétrie (c’est un composé asymétrique), le trans -diméthyl-1,2cyclopropane (formule 64) comporte un axe de symétrie d’ordre 2 (C2), le perhydrotriphénylène (formules 65 et 66) comporte un axe de symétrie d’ordre 3 (C3) et trois axes de symétrie d’ordre 2, perpendiculaires au premier. Ces composés (formules 63, 64, 65) ne sont pas superposables à leurs images par rapport à un plan; ils sont donc doués de chiralité. Pour reconnaître si une molécule est chirale, il suffit de vérifier l’absence des éléments de symétrie suivants: plan de symétrie, centre de symétrie et axe impropre de symétrie Sn (combinaison d’une rotation de 2 神/n et d’une symétrie par rapport à un plan perpendiculaire à l’axe de rotation). Ainsi, le spiranne (formule 67), dépourvu de plan et de centre de symétrie, n’est pas chiral à cause de la présence d’un axe de symétrie S4.Divers aspects de la chiralitéStructures chiralesCertains composés chiraux ont un carbone ou un soufre asymétrique et sont donc caractérisés par un pouvoir rotatoire (cf. supra , Carbone asymétrique ). On connaît maintenant de nombreux autres atomes asymétriques. Par exemple des phosphines (formule 68) ont été dédoublées. Du fait que l’azote incorporé dans des petits cycles ne s’inverse pas, l’aziridine (formule 69) optiquement active a été isolée. Un métal de transition tel que le manganèse (Mn), le fer (Fe) ou le chrome (Cr) peut jouer le rôle d’un centre asymétrique. C’est ainsi que le complexe métallique obtenu avec le fer (formule 70) a été dédoublé. Les cas d’atropo-isomérie (cf. supra , Atropo-isomérie ) se rencontrent de plus en plus fréquemment, donnant naissance à des énantiomères stables du fait d’empêchement de la rotation autour de liaisons C 漣C, comme pour certaines benzophénones (formule 71) ou l’hexahélicène (formule 72). Cette dernière molécule ne peut exister sous la conformation plane habituelle pour un système aromatique, à cause d’interactions entre les noyaux terminaux; une conformation très stable en hélice est alors adoptée. Toute la série homologue jusqu’au composé comportant 13 noyaux aromatiques est maintenant connue grâce aux travaux de R. H. Martin. Ces composés ont des pouvoirs rotatoires spécifiques parmi les plus élevés pour des composés organiques, comme celui de l’hexahélicène, 3 6000.Une double liaison C=C de stéréochimie trans ne peut être incorporée dans un cycle hexagonal ou pentagonal. Le cyclohexène, par exemple, possède une double liaison cis . Le cycle à 8 chaînons est le cycle le plus petit capable d’incorporer une double liaison trans . Le trans -cyclooctène est toutefois une molécule qui prend une conformation tendue (formule 73); celle-ci est chirale et ne peut s’inverser en son antipode. En effet, le trans -cyclooctène a effectivement été dédoublé en ses énantiomères par Cope. Si la taille du cycle croît, il devient impossible d’isoler des énantiomères par suite de la flexibilité de la molécule qui rend facile sa racémisation. Un cas très particulier de carbone asymétrique est celui où un composé tel que R 漣CH2R est rendu chiral par introduction d’un deutérium pour conduire à R 漣 CHDR . De tels centres asymétriques sont précieux pour établir la stéréochimie et le mécanisme de réactions enzymatiques. Cornforth et Arigoni ont réussi à préparer des groupes méthyle chiraux en mettant sur le même carbone hydrogène, deutérium et tritium. Ainsi, l’acide acétique chiral de configuration connue, CHDTC2H, a été synthétisé. Des méthodes biochimiques sont nécessaires pour évaluer pureté optique et configuration d’un tel composé, toujours préparé en quantité minime à cause de la radioactivité du tritium.Nomenclature de la configuration absolueLa nomenclature R , S proposée par Cahn, Ingold et Prelog est maintenant adoptée très généralement. On définit pour un carbone asymétrique Cabcd une priorité décroissante des groupes a , b , c et d . On imagine a , b , c , disposés sur un volant d’axe d . Le sigle R correspond à une séquence a , b , c distribuée dans le sens des aiguilles d’une montre (fig.1). La configuration S correspond au cas contraire. Un bon classement des groupes est essentiel pour une utilisation correcte de la méthode. Le classement, par numéro atomique décroissant, des atomes directement liés au centre asymétrique n’est pas valable entre chaînes carbonées (cf. supra le principe de la nomenclature, dans Carbone asymétrique ).Pour lever l’indétermination entre deux groupes, par exemple 漣CH3 et 漣CH2CH3, on poursuit la comparaison dans les chaînes carbonées. Ainsi, 漣CH3 est à considérer comme un carbone lié à trois hydrogènes, 漣CH2CH3 est un carbone lié à deux hydrogènes et un carbone. La comparaison de C (H, H, H ) et C (H, H, C ) permet de lever l’indétermination, l’atome C ayant priorité sur un hydrogène H . Le classement donne 漣CH2CH3 礪 漣CH3. Si les groupes carbonés sont plus complexes et ne permettent pas une levée rapide de l’indétermination, on continue la progression à l’intérieur des groupes et la comparaison relative jusqu’à levée de l’indétermination. En cas de ramification dans un groupe, on poursuit la progression en choisissant la branche prioritaire, selon les règles R , S . Par exemple, sur cette base:
conduit à un mélange en quantités inégales du nitrile tartrique racémique et du nitrile tartrique méso; on considère que l’intermédiaire est le mononitrile racémique N 令C 漣CHOH 漣CHO, ce qui ramène au type précédemment exposé.3. Chiralité et synthèse asymétriqueDurant les dix dernières années, de grands progrès ont été accomplis dans les méthodes d’études des structures chirales. Celles-ci sont importantes car elles interviennent dans beaucoup de processus biologiques. La majorité des molécules d’intérêt biologique sont chirales (polypeptides, enzymes, stéroïdes, prostaglandines, etc.). Les plus simples d’entre elles peuvent être obtenues par synthèse totale, mais seul l’énantiomère ayant la configuration absolue désirée (cf. supra , fig. 1) doit être sélectivement préparé.Le terme chiralité est dérivé du nom grec kheir ( 﨑﨎晴福, main) et caractérise deux objets qui, comme une main droite et une main gauche, sont différents tout en paraissant similaires. La distinction ne réside pas dans des dimensions ou des formes respectives. Une main gauche et une main droite ne peuvent être amenées en superposition, elles se déduisent l’une de l’autre par une symétrie par rapport à un plan. Presque tous les constituants chimiques des systèmes vivants sont chiraux: polypeptides, protéines, glucides, acides nucléiques; la chiralité est une des caractéristiques des organismes vivants. C’est, toutefois, à une date relativement récente (1950-1960) que ce terme utilisé par le physicien lord Kelvin au début du siècle a été introduit en chimie par Cahn, Ingold, Prelog, Mislow. Il devenait, en effet, indispensable de disposer d’un mot caractérisant des espèces moléculaires les plus variées qui peuvent exister sous forme de deux stéréo-isomères, image l’un de l’autre par rapport à un plan miroir. Le mot chiralité ne fait pas appel à des considérations de symétrie, il s’applique à tout composé pouvant avoir une image non superposable, appelé composé antipode ou énantiomère . Par exemple, l’acide lactique (formule 63) est dépourvu d’éléments de symétrie (c’est un composé asymétrique), le trans -diméthyl-1,2cyclopropane (formule 64) comporte un axe de symétrie d’ordre 2 (C2), le perhydrotriphénylène (formules 65 et 66) comporte un axe de symétrie d’ordre 3 (C3) et trois axes de symétrie d’ordre 2, perpendiculaires au premier. Ces composés (formules 63, 64, 65) ne sont pas superposables à leurs images par rapport à un plan; ils sont donc doués de chiralité. Pour reconnaître si une molécule est chirale, il suffit de vérifier l’absence des éléments de symétrie suivants: plan de symétrie, centre de symétrie et axe impropre de symétrie Sn (combinaison d’une rotation de 2 神/n et d’une symétrie par rapport à un plan perpendiculaire à l’axe de rotation). Ainsi, le spiranne (formule 67), dépourvu de plan et de centre de symétrie, n’est pas chiral à cause de la présence d’un axe de symétrie S4.Divers aspects de la chiralitéStructures chiralesCertains composés chiraux ont un carbone ou un soufre asymétrique et sont donc caractérisés par un pouvoir rotatoire (cf. supra , Carbone asymétrique ). On connaît maintenant de nombreux autres atomes asymétriques. Par exemple des phosphines (formule 68) ont été dédoublées. Du fait que l’azote incorporé dans des petits cycles ne s’inverse pas, l’aziridine (formule 69) optiquement active a été isolée. Un métal de transition tel que le manganèse (Mn), le fer (Fe) ou le chrome (Cr) peut jouer le rôle d’un centre asymétrique. C’est ainsi que le complexe métallique obtenu avec le fer (formule 70) a été dédoublé. Les cas d’atropo-isomérie (cf. supra , Atropo-isomérie ) se rencontrent de plus en plus fréquemment, donnant naissance à des énantiomères stables du fait d’empêchement de la rotation autour de liaisons C 漣C, comme pour certaines benzophénones (formule 71) ou l’hexahélicène (formule 72). Cette dernière molécule ne peut exister sous la conformation plane habituelle pour un système aromatique, à cause d’interactions entre les noyaux terminaux; une conformation très stable en hélice est alors adoptée. Toute la série homologue jusqu’au composé comportant 13 noyaux aromatiques est maintenant connue grâce aux travaux de R. H. Martin. Ces composés ont des pouvoirs rotatoires spécifiques parmi les plus élevés pour des composés organiques, comme celui de l’hexahélicène, 3 6000.Une double liaison C=C de stéréochimie trans ne peut être incorporée dans un cycle hexagonal ou pentagonal. Le cyclohexène, par exemple, possède une double liaison cis . Le cycle à 8 chaînons est le cycle le plus petit capable d’incorporer une double liaison trans . Le trans -cyclooctène est toutefois une molécule qui prend une conformation tendue (formule 73); celle-ci est chirale et ne peut s’inverser en son antipode. En effet, le trans -cyclooctène a effectivement été dédoublé en ses énantiomères par Cope. Si la taille du cycle croît, il devient impossible d’isoler des énantiomères par suite de la flexibilité de la molécule qui rend facile sa racémisation. Un cas très particulier de carbone asymétrique est celui où un composé tel que R 漣CH2R est rendu chiral par introduction d’un deutérium pour conduire à R 漣 CHDR . De tels centres asymétriques sont précieux pour établir la stéréochimie et le mécanisme de réactions enzymatiques. Cornforth et Arigoni ont réussi à préparer des groupes méthyle chiraux en mettant sur le même carbone hydrogène, deutérium et tritium. Ainsi, l’acide acétique chiral de configuration connue, CHDTC2H, a été synthétisé. Des méthodes biochimiques sont nécessaires pour évaluer pureté optique et configuration d’un tel composé, toujours préparé en quantité minime à cause de la radioactivité du tritium.Nomenclature de la configuration absolueLa nomenclature R , S proposée par Cahn, Ingold et Prelog est maintenant adoptée très généralement. On définit pour un carbone asymétrique Cabcd une priorité décroissante des groupes a , b , c et d . On imagine a , b , c , disposés sur un volant d’axe d . Le sigle R correspond à une séquence a , b , c distribuée dans le sens des aiguilles d’une montre (fig.1). La configuration S correspond au cas contraire. Un bon classement des groupes est essentiel pour une utilisation correcte de la méthode. Le classement, par numéro atomique décroissant, des atomes directement liés au centre asymétrique n’est pas valable entre chaînes carbonées (cf. supra le principe de la nomenclature, dans Carbone asymétrique ).Pour lever l’indétermination entre deux groupes, par exemple 漣CH3 et 漣CH2CH3, on poursuit la comparaison dans les chaînes carbonées. Ainsi, 漣CH3 est à considérer comme un carbone lié à trois hydrogènes, 漣CH2CH3 est un carbone lié à deux hydrogènes et un carbone. La comparaison de C (H, H, H ) et C (H, H, C ) permet de lever l’indétermination, l’atome C ayant priorité sur un hydrogène H . Le classement donne 漣CH2CH3 礪 漣CH3. Si les groupes carbonés sont plus complexes et ne permettent pas une levée rapide de l’indétermination, on continue la progression à l’intérieur des groupes et la comparaison relative jusqu’à levée de l’indétermination. En cas de ramification dans un groupe, on poursuit la progression en choisissant la branche prioritaire, selon les règles R , S . Par exemple, sur cette base: Dans l’exemple 漣CH2Br 礪 漣C(CH3)3, la décision est obtenue par la comparaison C (H, H, Br) et C (C, C, C), Br étant prioritaire sur tous les atomes liés au carbone de 漣C(CH3)3. Quand un groupe est insaturé, des règles ont été élaborées. Une double liaison est remplacée par une structure hypothétique saturée: on répète à chaque extrémité de la double liaison l’atome qui est à l’autre extrémité, ces deux atomes étant ramenés à la tétracoordination par des atomes «fantômes» 0, supposés avoir un numéro atomique de zéro (formule 74, en exemple). Les structures alléniques sont considérées comme des tétraèdres déformés auxquels on applique les règles générales. Il est intéressant de mentionner que la nomenclature cis , trans pour les oléfines tend à être remplacée par la nomenclature plus rigoureuse E , Z . Dans celle-ci, on classe à chaque extrémité de la double liaison les groupes selon les règles précédentes, et on sélectionne le groupe prioritaire. Une disposition cis des groupes prioritaires est appelée Z (zusammen ), une disposition trans correspond à la configuration E (entgegen ). Ainsi, le chloro-2 pentène-2 (formule 75) possède la configuration E , Cl et CH2CH3 étant les deux groupes prioritaires à considérer.ProchiralitéUn carbone asymétrique RR CXY peut être créé à partir de divers précurseurs: par exemple, à partir d’un composé du type RR CX2 où un des groupes X est remplacé par Y. Une autre possibilité est la réaction d’addition d’un réactif A 漣B sur un composé insaturé RR C=W (W égal à O ou à CR Rt), comme dans la réduction de C6H5COCH3 en C6H5CHOHCH3. Les précurseurs RR CX2 et RR C=W sont appelés prochiraux (on vérifie aisément que des composés tels que RCX3 ou R2C=W ne sont pas des composés prochiraux). Si on considère un carbone tétraédrique RR CX2, les groupes X ont des caractéristiques très différentes selon la nature de R et R . Trois cas peuvent se présenter: Si R est égal à R , R et R chiraux ou non chiraux, un axe de symétrie fait correspondre un des groupes X à l’autre et ils sont équivalents . Si R est distinct de R (R et R non chiraux), les groupes X ne sont plus équivalents et ils sont reliés par un plan de symétrie; ils sont appelés énantiotopiques ; ils ne sont différenciables que par un phénomène ou un réactif chiral, car ils sont dans des environnements locaux énantiomères. Enfin, si R ou R est chiral, le plan de symétrie disparaît (la molécule étant chirale); les groupes X ne sont plus reliés par une opération de symétrie, ils sont donc intrinsèquement différents; ils sont appelés groupes diastéréotopiques ; ils sont en principe différenciables par tout phénomène ou réactif achiral. Ainsi, un groupe méthylène 漣CH2 漣 donne, en résonance magnétique nucléaire (R.M.N.) du proton 1H, un seul signal si les deux hydrogènes sont équivalents ou énantiotopiques et deux signaux pour des protons diastéréotopiques. Il est nécessaire de nommer les substituants énantiotopiques ou diastéréotopiques. Pour cela, on utilise les règles de classement de la nomenclature R , S et on rend arbitrairement prioritaire sur l’autre le groupe X que l’on désire nommer; on applique ensuite au pseudo-atome asymétrique ainsi formé les règles R , S . Si on trouve une distribution par priorité décroissante dans le sens des aiguilles d’une montre, le groupe X est appelé pro-R . Dans le cas contraire, il est appelé pro-S . Par exemple, sont indiqués les hydrogènes pro-R et pro-S , souvent nommés HR HS , dans le dihydronicotamide, qui fait partie du coenzyme NADH (dihydropyridine-nucléotide, formule 76). Cet agent réducteur biochimique délivre spécifiquement HR ou HS sur un substrat organique selon l’enzyme qui lui est associée.La nomenclature des faces d’une double liaison prochirale fait appel à des conventions légèrement différentes. Pour nommer une face de l’acide pyruvique CH3 漣CO 漣C2H, par exemple, qui a une fonction cétone prochirale, on classe les trois groupes liés au carbone cétonique =O 礪 C2H 礪 CH3. La face où est observée cette séquence dans le sens des aiguilles d’une montre est appelée re (rectus ), un sens inverse définit une face si (sinister ).Activités biologiques et chiralitéLes activités biologiques des molécules organiques sont extrêmement variées et sont très sensibles à leur stéréochimie. Très souvent, un énantiomère donné possède essentiellement toute l’activité biologique, le deuxième énantiomère étant inactif, ou ayant des propriétés biologiques nouvelles. Cette spécificité remarquable tient au fait que les récepteurs biologiques, si complexes soient-ils, sont eux-mêmes chiraux, car ils mettent en jeu des macromolécules à base de polypeptides ou de polysaccharides. Il est donc essentiel de contrôler la synthèse d’un composé à action thérapeutique, afin d’éviter de l’obtenir sous forme racémique. Quelques exemples peuvent servir à illustrer les relations entre la chiralité et l’activité biologique. Le (face=F0019 漣)-citronnellol (lévogyre) a une odeur de géranium, alors que le (+)-citronnellol (dextrogyre) a une odeur d’huile de citronnelle. Il a été rigoureusement établi que cette différence d’odeur entre énantiomères n’était pas due à des traces d’impuretés variables selon l’énantiomère considéré. Le (face=F0019 漣)-menthol possède l’odeur mentholée bien connue alors que le (+)-menthol est inodore ou a une odeur désagréable (selon les individus). L’asparagine est un acide aminé important: un de ses antipodes (D) a un goût sucré alors que l’autre (L) a un goût amer. Les acides aminés naturels se présentent toujours, à de rares exceptions près, sous la configuration absolue L. La configuration des 見-aminoacides L est représentée par la projection de Fischer (formule 77) où les liaisons horizontales C 漣NH2 et C 漣H sont en avant et les liaisons verticales situées vers l’arrière. Remarquons que la nomenclature R , S ne permet pas d’attribuer une configuration à la famille: en effet pour R égal à CH3, on a la (S )-alanine, mais si R est égal à CH2 漣S 漣CH3, l’aminoacide est la (R )-cystéine. Un enchaînement d’aminoacides L du type NHCH(R)CONHCH(R )CONHCH (R )CO... conduit à des polypeptides. Par suite des liaisons hydrogènes intramoléculaires pouvant s’établir entre NH et CO d’aminoacides différents, la macromolécule prend une structure en hélice (hélice 見). La chiralité des composés monomères conduit à une chiralité conformationnelle, une hélice étant chirale. On retrouve un phénomène analogue dans les macromolécules encore plus complexes que sont les acides nucléiques: deux macromolécules, ayant un certain nombre de constituants chiraux, s’apparient par des réseaux de liaisons hydrogènes en donnant une double hélice.Divers types de synthèse asymétrique selon l’auxiliaire chiralLa préparation d’un énantiomère pur peut se faire par deux méthodes, qui font chacune appel à un auxiliaire chiral Z. Si on dispose du composé sous forme d’un mélange racémique (énantiomères R et S en quantités égales), il est possible de séparer l’antipode désiré, R (ou S ) grâce à un dédoublement . Cette opération consiste à transformer le mélange des énantiomères R , S en un mélange de diastéréo-isomères ZR , ZS , en principe séparables par les méthodes physiques habituelles (cristallisation, distillation, etc.). La liaison entre Z et R (ou S ) doit autoriser un clivage pour régénérer l’énantiomère désiré R (ou S ). Dans le meilleur des cas, en théorie, on isole 50 p. 100 de R (ou de S ) par rapport au mélange initial; il est nécessaire de recycler Z et de racémiser S (ou R ) quand cela est possible.La synthèse asymétrique (deuxième méthode) consiste à synthétiser directement l’énantiomère R (ou S ) à partir d’un substrat prochiral. Il est encore nécessaire d’introduire dans le système un auxiliaire chiral Z. En théorie, on peut obtenir 100 p. 100 d’un énantiomère à partir de la matière première prochirale.Évaluation de l’efficacité d’une synthèse asymétriquePendant longtemps, la composition énantiomérique d’un composé a été déduite de mesures polarimétriques. La pureté optique P est définie par le rapport ( 見)observé/( 見)max, où ( 見)max est le pouvoir rotatoire spécifique maximal correspondant au composé complètement dédoublé et ( 見)observé est le pouvoir rotatoire spécifique de l’échantillon considéré, mesuré dans les mêmes conditions. Il est facile de voir que :
Dans l’exemple 漣CH2Br 礪 漣C(CH3)3, la décision est obtenue par la comparaison C (H, H, Br) et C (C, C, C), Br étant prioritaire sur tous les atomes liés au carbone de 漣C(CH3)3. Quand un groupe est insaturé, des règles ont été élaborées. Une double liaison est remplacée par une structure hypothétique saturée: on répète à chaque extrémité de la double liaison l’atome qui est à l’autre extrémité, ces deux atomes étant ramenés à la tétracoordination par des atomes «fantômes» 0, supposés avoir un numéro atomique de zéro (formule 74, en exemple). Les structures alléniques sont considérées comme des tétraèdres déformés auxquels on applique les règles générales. Il est intéressant de mentionner que la nomenclature cis , trans pour les oléfines tend à être remplacée par la nomenclature plus rigoureuse E , Z . Dans celle-ci, on classe à chaque extrémité de la double liaison les groupes selon les règles précédentes, et on sélectionne le groupe prioritaire. Une disposition cis des groupes prioritaires est appelée Z (zusammen ), une disposition trans correspond à la configuration E (entgegen ). Ainsi, le chloro-2 pentène-2 (formule 75) possède la configuration E , Cl et CH2CH3 étant les deux groupes prioritaires à considérer.ProchiralitéUn carbone asymétrique RR CXY peut être créé à partir de divers précurseurs: par exemple, à partir d’un composé du type RR CX2 où un des groupes X est remplacé par Y. Une autre possibilité est la réaction d’addition d’un réactif A 漣B sur un composé insaturé RR C=W (W égal à O ou à CR Rt), comme dans la réduction de C6H5COCH3 en C6H5CHOHCH3. Les précurseurs RR CX2 et RR C=W sont appelés prochiraux (on vérifie aisément que des composés tels que RCX3 ou R2C=W ne sont pas des composés prochiraux). Si on considère un carbone tétraédrique RR CX2, les groupes X ont des caractéristiques très différentes selon la nature de R et R . Trois cas peuvent se présenter: Si R est égal à R , R et R chiraux ou non chiraux, un axe de symétrie fait correspondre un des groupes X à l’autre et ils sont équivalents . Si R est distinct de R (R et R non chiraux), les groupes X ne sont plus équivalents et ils sont reliés par un plan de symétrie; ils sont appelés énantiotopiques ; ils ne sont différenciables que par un phénomène ou un réactif chiral, car ils sont dans des environnements locaux énantiomères. Enfin, si R ou R est chiral, le plan de symétrie disparaît (la molécule étant chirale); les groupes X ne sont plus reliés par une opération de symétrie, ils sont donc intrinsèquement différents; ils sont appelés groupes diastéréotopiques ; ils sont en principe différenciables par tout phénomène ou réactif achiral. Ainsi, un groupe méthylène 漣CH2 漣 donne, en résonance magnétique nucléaire (R.M.N.) du proton 1H, un seul signal si les deux hydrogènes sont équivalents ou énantiotopiques et deux signaux pour des protons diastéréotopiques. Il est nécessaire de nommer les substituants énantiotopiques ou diastéréotopiques. Pour cela, on utilise les règles de classement de la nomenclature R , S et on rend arbitrairement prioritaire sur l’autre le groupe X que l’on désire nommer; on applique ensuite au pseudo-atome asymétrique ainsi formé les règles R , S . Si on trouve une distribution par priorité décroissante dans le sens des aiguilles d’une montre, le groupe X est appelé pro-R . Dans le cas contraire, il est appelé pro-S . Par exemple, sont indiqués les hydrogènes pro-R et pro-S , souvent nommés HR HS , dans le dihydronicotamide, qui fait partie du coenzyme NADH (dihydropyridine-nucléotide, formule 76). Cet agent réducteur biochimique délivre spécifiquement HR ou HS sur un substrat organique selon l’enzyme qui lui est associée.La nomenclature des faces d’une double liaison prochirale fait appel à des conventions légèrement différentes. Pour nommer une face de l’acide pyruvique CH3 漣CO 漣C2H, par exemple, qui a une fonction cétone prochirale, on classe les trois groupes liés au carbone cétonique =O 礪 C2H 礪 CH3. La face où est observée cette séquence dans le sens des aiguilles d’une montre est appelée re (rectus ), un sens inverse définit une face si (sinister ).Activités biologiques et chiralitéLes activités biologiques des molécules organiques sont extrêmement variées et sont très sensibles à leur stéréochimie. Très souvent, un énantiomère donné possède essentiellement toute l’activité biologique, le deuxième énantiomère étant inactif, ou ayant des propriétés biologiques nouvelles. Cette spécificité remarquable tient au fait que les récepteurs biologiques, si complexes soient-ils, sont eux-mêmes chiraux, car ils mettent en jeu des macromolécules à base de polypeptides ou de polysaccharides. Il est donc essentiel de contrôler la synthèse d’un composé à action thérapeutique, afin d’éviter de l’obtenir sous forme racémique. Quelques exemples peuvent servir à illustrer les relations entre la chiralité et l’activité biologique. Le (face=F0019 漣)-citronnellol (lévogyre) a une odeur de géranium, alors que le (+)-citronnellol (dextrogyre) a une odeur d’huile de citronnelle. Il a été rigoureusement établi que cette différence d’odeur entre énantiomères n’était pas due à des traces d’impuretés variables selon l’énantiomère considéré. Le (face=F0019 漣)-menthol possède l’odeur mentholée bien connue alors que le (+)-menthol est inodore ou a une odeur désagréable (selon les individus). L’asparagine est un acide aminé important: un de ses antipodes (D) a un goût sucré alors que l’autre (L) a un goût amer. Les acides aminés naturels se présentent toujours, à de rares exceptions près, sous la configuration absolue L. La configuration des 見-aminoacides L est représentée par la projection de Fischer (formule 77) où les liaisons horizontales C 漣NH2 et C 漣H sont en avant et les liaisons verticales situées vers l’arrière. Remarquons que la nomenclature R , S ne permet pas d’attribuer une configuration à la famille: en effet pour R égal à CH3, on a la (S )-alanine, mais si R est égal à CH2 漣S 漣CH3, l’aminoacide est la (R )-cystéine. Un enchaînement d’aminoacides L du type NHCH(R)CONHCH(R )CONHCH (R )CO... conduit à des polypeptides. Par suite des liaisons hydrogènes intramoléculaires pouvant s’établir entre NH et CO d’aminoacides différents, la macromolécule prend une structure en hélice (hélice 見). La chiralité des composés monomères conduit à une chiralité conformationnelle, une hélice étant chirale. On retrouve un phénomène analogue dans les macromolécules encore plus complexes que sont les acides nucléiques: deux macromolécules, ayant un certain nombre de constituants chiraux, s’apparient par des réseaux de liaisons hydrogènes en donnant une double hélice.Divers types de synthèse asymétrique selon l’auxiliaire chiralLa préparation d’un énantiomère pur peut se faire par deux méthodes, qui font chacune appel à un auxiliaire chiral Z. Si on dispose du composé sous forme d’un mélange racémique (énantiomères R et S en quantités égales), il est possible de séparer l’antipode désiré, R (ou S ) grâce à un dédoublement . Cette opération consiste à transformer le mélange des énantiomères R , S en un mélange de diastéréo-isomères ZR , ZS , en principe séparables par les méthodes physiques habituelles (cristallisation, distillation, etc.). La liaison entre Z et R (ou S ) doit autoriser un clivage pour régénérer l’énantiomère désiré R (ou S ). Dans le meilleur des cas, en théorie, on isole 50 p. 100 de R (ou de S ) par rapport au mélange initial; il est nécessaire de recycler Z et de racémiser S (ou R ) quand cela est possible.La synthèse asymétrique (deuxième méthode) consiste à synthétiser directement l’énantiomère R (ou S ) à partir d’un substrat prochiral. Il est encore nécessaire d’introduire dans le système un auxiliaire chiral Z. En théorie, on peut obtenir 100 p. 100 d’un énantiomère à partir de la matière première prochirale.Évaluation de l’efficacité d’une synthèse asymétriquePendant longtemps, la composition énantiomérique d’un composé a été déduite de mesures polarimétriques. La pureté optique P est définie par le rapport ( 見)observé/( 見)max, où ( 見)max est le pouvoir rotatoire spécifique maximal correspondant au composé complètement dédoublé et ( 見)observé est le pouvoir rotatoire spécifique de l’échantillon considéré, mesuré dans les mêmes conditions. Il est facile de voir que : R et S représentant les quantités relatives des deux énantiomères. À partir de P, il est possible de calculer R et S (en pourcentage). On évalue maintenant la composition énantiomérique par des méthodes non polarimétriques; aussi le terme de pureté optique est-il remplacé par le terme d’excès énantiomérique (e.e. en abrégé), défini par:
R et S représentant les quantités relatives des deux énantiomères. À partir de P, il est possible de calculer R et S (en pourcentage). On évalue maintenant la composition énantiomérique par des méthodes non polarimétriques; aussi le terme de pureté optique est-il remplacé par le terme d’excès énantiomérique (e.e. en abrégé), défini par: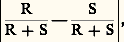 L’excès énantiomérique peut être mesuré par chromatographie gazeuse ou liquide-liquide avec phase stationnaire chirale. La résonance magnétique nucléaire, en présence d’un solvant chiral, est également un moyen commode d’analyser une composition d’énantiomères. Les méthodes chromatographiques sont des plus sensibles et permettent de travailler sur de très faibles quantités (de l’ordre du microgramme). L’excès énantiomérique d’aminoacides présents à l’état de traces dans les météorites a pu ainsi être examiné, dans l’espoir de détecter indirectement la présence d’organismes vivants à l’extérieur de la Terre (si e.e. 0). Jusqu’à présent, seule la composition racémique a été observée.L’efficacité d’une synthèse asymétrique est chiffrée par la pureté optique P du produit, exprimée en pourcentage, ou mieux par e.e. (%). Si l’auxiliaire chiral Z n’est pas énantiomériquement pur, il convient alors d’en tenir compte par une correction qui consiste habituellement à diviser P (%) ou e.e. (%) par la pureté optique de Z.Différents types de synthèse asymétriqueLa synthèse asymétrique peut prendre divers aspects, selon la manière dont est mis en œuvre l’auxiliaire chiral Z.Auxiliaire chiral Z lié au substrat prochiralCelui-ci peut être temporairement lié au substrat prochiral RR CX2 ou RR C=Y, par l’intermédiaire d’un groupe fonctionnel présent dans R ou R , par exemple; on aboutit ainsi au composé Z 漣R(R CX2) ou Z 漣R(R C=Y). Dans ces conditions, il y a disparition du plan de symétrie du carbone tétraédrique de RR CX2 ou de RR C=Y (plan de la double liaison), la molécule étant devenue chirale. Les deux groupes X et les deux faces de C=Y n’étant plus reliés par une opération de symétrie, ils sont donc différenciés et ont pris un caractère diastéréotopique. En conséquence, un réactif achiral convenablement choisi est, en principe, capable de réagir sélectivement avec un groupe X ou une face d’un carbone trigonal de ces composés intermédiaires Z 漣R(R CX2) ou Z 漣R(R C=Y). Z oriente le cours stérique de la réaction du substrat; aussi ce type de synthèse est-il parfois appelé synthèse asymétrique diastéréosélective. En exemple, la synthèse asymétrique de l’acide atrolactique C6H5(CH3)C(OH)C2H par action de l’iodure de méthyl-magnésium CH3MgI (organomagnésien), sur un ester de l’acide phénylglyoxylique C6H5COC2H. La saponification permet de séparer l’alcool chiral initial, symbolisé par (G, M, P)C 漣OH, de l’acide atrolactique formé. G, M et P représentent trois groupes de taille différente (G pour gros, M pour moyen et P pour petit). Prelog a pu corréler la configuration absolue de l’acide atrolactique formé avec celle de l’alcool chiral mis en jeu. La «règle de Prelog» a été très utilisée pour établir la configuration d’alcools chiraux, en estérifiant ceux-ci par l’acide phénylglyoxylique et en examinant la configuration absolue de l’acide atrolactique, issu de la réaction précédente. Prelog a supposé que les phénylglyoxylates réagissaient avec l’organomagnésien en adoptant une conformation où les deux carbonyles étaient antiparallèles et la liaison C 漣G dans le plan de la fonction ester. Dans la conformation représentée (formule 78), seules les liaisons C 漣P et C 漣M sont hors du plan, l’organométallique attaquant par l’avant du plan, du côté du petit substituant. L’acide atrolactique (S ) résulte de cette réaction. La règle de Prelog est très utile pour prévoir des expériences: elle ne comporte pratiquement pas d’exception. Mais telle qu’elle est formulée, elle ne représente sans doute pas exactement le mécanisme de la réaction.Réactif chiral réagissant sur un substrat prochiralUn deuxième mode de synthèse asymétrique consiste à utiliser un réactif chiral qui comporte l’auxiliaire Z et qui réagit sur un substrat prochiral. Le réactif chiral peut sélectionner un des groupes X de RR CX2 ou l’une des faces de RR C=Y. En effet, il faut à chaque fois considérer qu’il y a compétition entre deux états de transition diastéréo-isomères (et non énantiomères) qui ont donc des énergies différentes. Par exemple, la réduction asymétrique de l’acétophénone par l’organomagnésien (S ) 漣CH3CH (C2H5) 漣CH2MgBr conduit au (R ) 漣 phénylméthylcarbinol. Le déroulement stérique de la réaction s’explique aisément en sachant qu’il y a un transfert d’hydrure de l’organomagnésien vers la cétone, par un mécanisme cyclique. Dans le cas présent, l’état de transition privilégié est celui où le minimum d’interaction stérique est obtenu en plaçant le phényle en face du méthyle de l’agent réducteur (formule 79). Les synthèses où l’une des faces énantiotopiques est sélectivement transformée sont parfois nommées synthèses asymétriques énantiosélectives. Le réactif chiral n’est pas nécessairement chimique, il peut être un système enzymatique, comme la levure de boulanger, qui permet de réduire en milieu aqueux C6H5COCH2OH en (R ) 漣C6H5CHOHCH2OH avec une stéréosélectivité parfaite.La catalyse asymétrique est, par nature, complexe puisqu’elle met en jeu un cycle de réaction amenant la régénération du catalyseur: il est difficile, en général, de déceler l’étape clé où l’induction asymétrique s’exerce. Cependant, dans deux types de catalyse, les progrès ont été spectaculaires.Il est connu que l’aldolisation peut être catalysée par un acide ou une base. Des chercheurs des sociétés Schering et Hoffmann-La Roche ont montré que la tricétone (formule 80) est cyclisée asymétriquement en cétol (formule 81) par une quantité catalytique d’un 見-aminoacide, la (S) 漣proline. Le cétol est transformé, par déshydratation, en une cétone éthylénique (formule 82), obtenue avec 95 p. 100 de pureté optique, qui est une matière première pour la synthèse totale des stéroïdes.Une autre catalyse, celle par des complexes organométalliques , permet de réaliser de nombreuses synthèses organiques: ainsi, des complexes du rhodium RhClL2S, où L représente une phosphine, S une molécule de solvant et Cl le chlore, sont de puissants catalyseurs d’hydrogénation d’oléfines. Découverts par Wilkinson, ils ont été récemment modifiés par l’introduction de phosphines chirales. Des stéréosélectivités remarquables (95-99 p. 100) ont été obtenues avec ces systèmes dans la synthèse asymétrique d’acides 見-aminés N-acétylés R CH(NHCOCH3)C2H par réduction de RCH=C(NHCOCH3)C2H. Un dernier exemple de catalyse asymétrique est la synthèse de l’acide chrysanthémique, composant important d’insecticide. Seul l’antipode (formule 83) est actif; il a pu être obtenu par addition asymétrique du carbène CHC2CH2CH3 sur (CH3)2C=CH 漣CH=C(CH3)2. La réaction consiste à décomposer asymétriquement 2CHC2CH2CH3 par un complexe chiral de cuivre en présence du diène. Il est vraisemblable que le carbène formé par élimination d’azote reste attaché au complexe de cuivre et réagit en tant que tel sur le diène. L’acide chrysanthémique est obtenu par saponification de l’ester éthylique formé dans la réaction précédente (formule 83).La stéréochimie organique, après une évolution continue dans les années 1950-1970, s’appuie maintenant sur des bases solides. Les structures, les conformations ou les configurations sont bien établies pour de nombreux composés, grâce aux méthodes physico-chimiques modernes. Le langage s’est précisé; la nomenclature R , S s’est imposée. Des composés chiraux n’ayant pas uniquement des carbones asymétriques sont de plus en plus fréquemment découverts. La stéréochimie de systèmes macromoléculaires d’intérêt biologique (mais non détaillés dans cet article) commence à être connue. La radiocristallographie a permis d’observer la conformation de certaines enzymes, conduisant à des hypothèses sur la nature des sites actifs qui induisent presque toujours des réactions hautement stéréosélectives.Les progrès de la synthèse asymétrique ont permis, dans certains cas, la préparation très sélective d’un énantiomère. Il est certain que la synthèse asymétrique des molécules chirales prendra une importance croissante. En effet, la synthèse de composés biologiquement actifs, d’additifs alimentaires, de constituants de parfums... nécessite le plus souvent, à un stade ou à un autre, une matière première chirale, qui pourrait avantageusement être obtenue par synthèse asymétrique.
L’excès énantiomérique peut être mesuré par chromatographie gazeuse ou liquide-liquide avec phase stationnaire chirale. La résonance magnétique nucléaire, en présence d’un solvant chiral, est également un moyen commode d’analyser une composition d’énantiomères. Les méthodes chromatographiques sont des plus sensibles et permettent de travailler sur de très faibles quantités (de l’ordre du microgramme). L’excès énantiomérique d’aminoacides présents à l’état de traces dans les météorites a pu ainsi être examiné, dans l’espoir de détecter indirectement la présence d’organismes vivants à l’extérieur de la Terre (si e.e. 0). Jusqu’à présent, seule la composition racémique a été observée.L’efficacité d’une synthèse asymétrique est chiffrée par la pureté optique P du produit, exprimée en pourcentage, ou mieux par e.e. (%). Si l’auxiliaire chiral Z n’est pas énantiomériquement pur, il convient alors d’en tenir compte par une correction qui consiste habituellement à diviser P (%) ou e.e. (%) par la pureté optique de Z.Différents types de synthèse asymétriqueLa synthèse asymétrique peut prendre divers aspects, selon la manière dont est mis en œuvre l’auxiliaire chiral Z.Auxiliaire chiral Z lié au substrat prochiralCelui-ci peut être temporairement lié au substrat prochiral RR CX2 ou RR C=Y, par l’intermédiaire d’un groupe fonctionnel présent dans R ou R , par exemple; on aboutit ainsi au composé Z 漣R(R CX2) ou Z 漣R(R C=Y). Dans ces conditions, il y a disparition du plan de symétrie du carbone tétraédrique de RR CX2 ou de RR C=Y (plan de la double liaison), la molécule étant devenue chirale. Les deux groupes X et les deux faces de C=Y n’étant plus reliés par une opération de symétrie, ils sont donc différenciés et ont pris un caractère diastéréotopique. En conséquence, un réactif achiral convenablement choisi est, en principe, capable de réagir sélectivement avec un groupe X ou une face d’un carbone trigonal de ces composés intermédiaires Z 漣R(R CX2) ou Z 漣R(R C=Y). Z oriente le cours stérique de la réaction du substrat; aussi ce type de synthèse est-il parfois appelé synthèse asymétrique diastéréosélective. En exemple, la synthèse asymétrique de l’acide atrolactique C6H5(CH3)C(OH)C2H par action de l’iodure de méthyl-magnésium CH3MgI (organomagnésien), sur un ester de l’acide phénylglyoxylique C6H5COC2H. La saponification permet de séparer l’alcool chiral initial, symbolisé par (G, M, P)C 漣OH, de l’acide atrolactique formé. G, M et P représentent trois groupes de taille différente (G pour gros, M pour moyen et P pour petit). Prelog a pu corréler la configuration absolue de l’acide atrolactique formé avec celle de l’alcool chiral mis en jeu. La «règle de Prelog» a été très utilisée pour établir la configuration d’alcools chiraux, en estérifiant ceux-ci par l’acide phénylglyoxylique et en examinant la configuration absolue de l’acide atrolactique, issu de la réaction précédente. Prelog a supposé que les phénylglyoxylates réagissaient avec l’organomagnésien en adoptant une conformation où les deux carbonyles étaient antiparallèles et la liaison C 漣G dans le plan de la fonction ester. Dans la conformation représentée (formule 78), seules les liaisons C 漣P et C 漣M sont hors du plan, l’organométallique attaquant par l’avant du plan, du côté du petit substituant. L’acide atrolactique (S ) résulte de cette réaction. La règle de Prelog est très utile pour prévoir des expériences: elle ne comporte pratiquement pas d’exception. Mais telle qu’elle est formulée, elle ne représente sans doute pas exactement le mécanisme de la réaction.Réactif chiral réagissant sur un substrat prochiralUn deuxième mode de synthèse asymétrique consiste à utiliser un réactif chiral qui comporte l’auxiliaire Z et qui réagit sur un substrat prochiral. Le réactif chiral peut sélectionner un des groupes X de RR CX2 ou l’une des faces de RR C=Y. En effet, il faut à chaque fois considérer qu’il y a compétition entre deux états de transition diastéréo-isomères (et non énantiomères) qui ont donc des énergies différentes. Par exemple, la réduction asymétrique de l’acétophénone par l’organomagnésien (S ) 漣CH3CH (C2H5) 漣CH2MgBr conduit au (R ) 漣 phénylméthylcarbinol. Le déroulement stérique de la réaction s’explique aisément en sachant qu’il y a un transfert d’hydrure de l’organomagnésien vers la cétone, par un mécanisme cyclique. Dans le cas présent, l’état de transition privilégié est celui où le minimum d’interaction stérique est obtenu en plaçant le phényle en face du méthyle de l’agent réducteur (formule 79). Les synthèses où l’une des faces énantiotopiques est sélectivement transformée sont parfois nommées synthèses asymétriques énantiosélectives. Le réactif chiral n’est pas nécessairement chimique, il peut être un système enzymatique, comme la levure de boulanger, qui permet de réduire en milieu aqueux C6H5COCH2OH en (R ) 漣C6H5CHOHCH2OH avec une stéréosélectivité parfaite.La catalyse asymétrique est, par nature, complexe puisqu’elle met en jeu un cycle de réaction amenant la régénération du catalyseur: il est difficile, en général, de déceler l’étape clé où l’induction asymétrique s’exerce. Cependant, dans deux types de catalyse, les progrès ont été spectaculaires.Il est connu que l’aldolisation peut être catalysée par un acide ou une base. Des chercheurs des sociétés Schering et Hoffmann-La Roche ont montré que la tricétone (formule 80) est cyclisée asymétriquement en cétol (formule 81) par une quantité catalytique d’un 見-aminoacide, la (S) 漣proline. Le cétol est transformé, par déshydratation, en une cétone éthylénique (formule 82), obtenue avec 95 p. 100 de pureté optique, qui est une matière première pour la synthèse totale des stéroïdes.Une autre catalyse, celle par des complexes organométalliques , permet de réaliser de nombreuses synthèses organiques: ainsi, des complexes du rhodium RhClL2S, où L représente une phosphine, S une molécule de solvant et Cl le chlore, sont de puissants catalyseurs d’hydrogénation d’oléfines. Découverts par Wilkinson, ils ont été récemment modifiés par l’introduction de phosphines chirales. Des stéréosélectivités remarquables (95-99 p. 100) ont été obtenues avec ces systèmes dans la synthèse asymétrique d’acides 見-aminés N-acétylés R CH(NHCOCH3)C2H par réduction de RCH=C(NHCOCH3)C2H. Un dernier exemple de catalyse asymétrique est la synthèse de l’acide chrysanthémique, composant important d’insecticide. Seul l’antipode (formule 83) est actif; il a pu être obtenu par addition asymétrique du carbène CHC2CH2CH3 sur (CH3)2C=CH 漣CH=C(CH3)2. La réaction consiste à décomposer asymétriquement 2CHC2CH2CH3 par un complexe chiral de cuivre en présence du diène. Il est vraisemblable que le carbène formé par élimination d’azote reste attaché au complexe de cuivre et réagit en tant que tel sur le diène. L’acide chrysanthémique est obtenu par saponification de l’ester éthylique formé dans la réaction précédente (formule 83).La stéréochimie organique, après une évolution continue dans les années 1950-1970, s’appuie maintenant sur des bases solides. Les structures, les conformations ou les configurations sont bien établies pour de nombreux composés, grâce aux méthodes physico-chimiques modernes. Le langage s’est précisé; la nomenclature R , S s’est imposée. Des composés chiraux n’ayant pas uniquement des carbones asymétriques sont de plus en plus fréquemment découverts. La stéréochimie de systèmes macromoléculaires d’intérêt biologique (mais non détaillés dans cet article) commence à être connue. La radiocristallographie a permis d’observer la conformation de certaines enzymes, conduisant à des hypothèses sur la nature des sites actifs qui induisent presque toujours des réactions hautement stéréosélectives.Les progrès de la synthèse asymétrique ont permis, dans certains cas, la préparation très sélective d’un énantiomère. Il est certain que la synthèse asymétrique des molécules chirales prendra une importance croissante. En effet, la synthèse de composés biologiquement actifs, d’additifs alimentaires, de constituants de parfums... nécessite le plus souvent, à un stade ou à un autre, une matière première chirale, qui pourrait avantageusement être obtenue par synthèse asymétrique.
Encyclopédie Universelle. 2012.